INTERACTIONS OF COMPLEMENT WITH HIV.
Complement Activation by HIV
Similarly to other pathogens, HIV infection results in activation of the complement system. HIV triggers the classical pathway, even in the absence of HIV-specific antibodies.
Direct binding of the viral envelope protein gp41 to C1q causes this activation mediating the deposition of C3 fragments on the viral surface [18,19]. By epitope mapping three C1q-binding sites on gp41 were identified, which depend on hydrophobic interactions [20]. After seroconversion, HIV-specific antibodies further enhance complement activation [21]. In addition, MBL, the triggering molecule of the lectin pathway, was also described to bind HIV [22]. MBL interacts with the virus via high mannose carbohydrates on gp120 and the interaction of MBL with HIV depends on sialysation [22].Inactivation of HIV Through Complement-Mediated Lysis
Several antibodies that induce complement-mediated lysis (CoML) of HIV have been described [23,24]. In vivo, such CoML-inducing antibodies are supposed to contribute to the control of the viral spread during the acute phase of infection. Mainly non-neutralizing antibodies seem to dominate this process [25,26]. The responses are thought to be found in the chronic phase of infection, too [25-29] (Fig. 1A). However, substantial amounts of the virus seem to be resistant against the lytic attacks by the complement system [20,30,31].
Responsible for this intrinsic resistance of HIV against human complement are membrane proteins derived from the human cells, which are acquired by the virus during the budding process [32]. Among them are RCAs such as CD46 (MCP), CD55 (DAF) or CD59, which down-regulate complement activation at several stages of the cascade [33-35]. In addition, HIV can bind fH, an RCA in fluid phase, which further favours the protection of the virus against lysis by the complement system [36-39].
The crucial role of fH for protection of the virus is evident, since incubation of HIV with fH-depleted sera results in up to 80% of complementdependent virolysis in the presence of HIV-specific antibodies [40].Similar to fH-depleted sera, which promote CoML, fH-derived peptide is able to enhance C3 deposition on HIV-infected cells by inhibition of fH deposition on HIV and thus to induce virolysis [41].
(A) Complement-mediated lysis (B) masking of epitopes
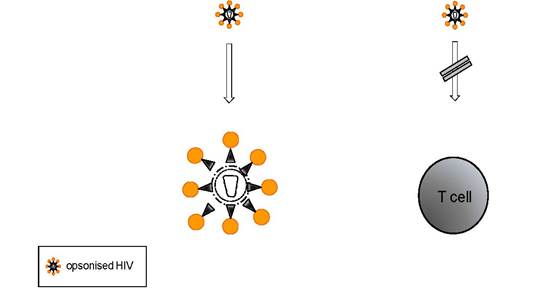
Figure 1: Inactivation of HIV by complement: (A) The complement system contributes to the control of the viral burden in infected individuals. In addition (B), opsonisation of the viral surface with C3-fragments may mask viral epitopes crucial for the adherence to target cells and/or the fusion process.
Inactivation Through Opsonization
Non-neutralizing Abs produced at early stages of infection is correlated with reduced infectivity, which seems to depend on complement [26]. Although some lysis was observed (see above), the covering of HIV with C3- fragments, i.e. opsonization, may also play an important role in controlling viral replication. In vitro data indicate that the masking of viral epitopes by deposition of C3-fragments on the viral envelope reduces the infectivity of complement-receptor-negative T cells [42,43]. This neutralization mechanism has been described for other viruses, too, [44] and may contribute, at least in part, to lower viral loads during the acute HIV infection (Fig. 1B).
Inactivation Through Phagocytosis
The coating with antibodies and complement tags HIV for uptake and destruction by phagocytes, such as dendritic cells (DCs), monocytes/macrophages or polymorphonuclear granulocytes [45]. Phagocytic cells internalize immune complexed viruses via their Fc and complement receptors (CRs), which upon engagement trigger uptake and subsequent degradation. In CR-mediated phagocytosis, all cell-bound C3-fragments act as opsonins and favor binding to the phagocyte via CRs [46].
In as much phagocytosis contributes to the reduction in viral loads of HIV-infected individuals is not completely elucidated yet. Defects in both complement and Fc receptor-dependent phagocytosis of macrophages and neutrophils are reported during disease progression [47-51].Complement-Mediated Enhancement of HIV Infection
Without intervention HIV remains resistant to human serum. Due to the protection mechanisms, described above, opsonized HIV accumulates in several compartments of the host, such as blood, lymphatic tissue (LT), brain, mother’s milk, seminal fluid or mucosal surfaces and can interact with CR expressing cells. The infectivity of opsonized virions may be rescued, or even enhanced, through exploitation of CRs on target and bystander cells (Fig. 2). By these mechanisms, the infection, dissemination and establishment of viral reservoirs is facilitated, as outlined below.
Complement-Mediated Enhancement of HIV Infection in CIS
Complement-mediated enhancement of infection is the topic of various reviews [20,52-56]. Such an enhancement in cis is reported after cross-linking of the CR1 on T cells with antibodies against this CR [57] (Fig. 2A). Similarly, aggregated C3b enhances transcription of viral genes in HIV-infected cells in vitro and in T lymphocytes isolated from HIV-infected individuals [57]. However, only a small subset of CD4+ T cells express CR1. Thus, the in vivo role of the CR1 for an infection of T cells in cis remains to be elucidated. Enhancement of infection has further been observed in CR2+ cell lines of T [58-61] or B cell origin [62,63] as well as in primary cultures of B lymphocytes [64,65] and syncytio-trophoblasts [66]. In most of these in vitro studies, simultaneous expression of CD4 is required for productive infection, although some studies demonstrated CD4-independent infection of CR2+ cells [67,68] (Fig. 2A).
Not only CR2, but also CR3 mediates enhancement HIV infection in cis, since on the one hand viral particles acquire ICAM-1 and CR3 during the budding process from monocytic cells [32], on the other hand complement-coated HIV interacts with CR3+ cells [69].
The host-derived ICAM-1 glycoprotein on the viral surface was shown to be biologically active and enhance viral infectivity in a CD4 independent manner [70], probably due to interaction with its counter-receptors LFA-1 and CR3 on the surface of the target cell. Incorporation of the intercellular adhesion molecule ICAM-1 into viral particles increased virus infectivity on peripheral blood mononuclear cells (PBMCs) by two- to sevenfold [71]. Treatment of target PBMCs with an antibody against LFA-1, a main ICAM-1 receptor, was able to block the ICAM-1-mediated enhancement. Adhesion between ICAM-1 on the target cell and CR3 on the virions could also mediate enhancement of viral infectivity, even without opsonising complement fragments on the viral surface due to C3-homolgy regions in gp41 [37]. Experiments with monocytic cells have shown that viral replication is increased, when monocytic cells were infected with iC3b-opsonized viral particles [72]. Simultaneous binding of iC3b to CR3 and of gp120 to CD4 and CCR5 has been suggested. Latently infected monocytes may be activated during a secondary infection by binding of opsonized particles or immune complexes to CR3, thereby inducing NFkB translocation and viral transcription.(A) Enhancement of infection in cis (B) Enhancement of infection in trans
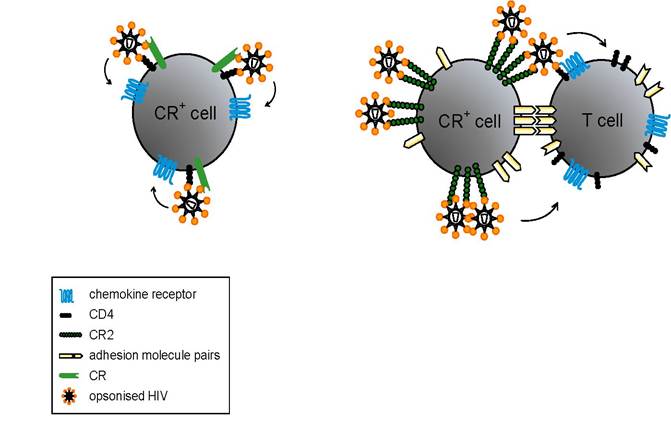
Figure 2: Complement-mediated enhancement of infection: (A) Binding of opsonized virus to CRs expressed on the target cells favours the infection of CD4- and chemokine receptor-positive cells. (B) Trapping of HIV on CD4- and chemokine receptor-negative cells via CRs promotes the transfer of HIV to T cells.
Such activated monocytes release high amounts of viral particles and secrete inflammatory cytokines [73]. As mentioned above CR3 is present on a large variety of cell types. Many aspects of the interaction between HIV and CR3 still remain to be elucidated. One example is the presence of CR3 on epithelial cells, which has been suggested to be involved in HIV transmission through rectal and cervicovaginal mucosa [74].
Another open question touches the FcR- and CR-mediated internalization of opsonized HIV which may profoundly affects intracellular sorting and antigen presentation [75].Complement-Mediated Enhancement of HIV Infection in Trans
CR1 seems to play a role for the pathogenesis of complement-opsonized HIV by contributing to the infection in trans. Immune-complexed HIV adhere to erythrocytes of infected individuals through CR1-C3b interactions [76,77] and transfer the virus - at least in vitro- to T cells (Fig. 2A). Additionally, CR1 on erythrocytes might be important for processing of complement fragments on the surface of HIV to generate C3dg [78]. This in turn can mediate the trapping of viral particles to CR2+ cells, thus enhancing the infection in trans [12] (Fig. 2B). In line with this observation, trapping of complement-coated virions to CR2+ cells has been shown with peripheral B lymphocytes isolated from HIV-infected patients [79] and in ex vivo studies for follicular dendritic cells (FDCs) [80-82]. The FDCs in the LT represents by far the largest viral reservoir in the body; up to 90 % of viral particles are extracellularly bound to FDCs [83,84]. Since FDCs express substantial amounts of CR1 and CR2 as well as lesser quantities of CR3 [85], they interact with all major C3 degradation products. In vitro, FDC-trapping of HIV is dependent on complement and on HIV-specific antibodies [81,86], although, accumulation of retroviruses in LT reservoirs is already observable in the acute phase of infection [87-90], when Abs are absent and HIV is covered with complement-fragments only [91]. Trapped virions were shown to remain infectious for T cells migrating through GC even in the presence of neutralising antibodies [92]. Although HAART reduces the pool of FDCS-associated HIV by over 2500-fold, viral RNA can still be detected in some GC [93].
In an ex vivo study with tonsilar specimen of a presymptomatic HIV-positive patients, CR2 was identified as the main ligand for in vivo complement-coated virus [80,81].
Monoclonal antibodies blocking C3d-CR2 interaction but not antibodies against CR1 or CR3 could detach the main part of trapped virions from the FDC network. Since the same antibodies could detach virus from tonsillar cell suspensions of patients receiving HAART, trapping mechanisms do not seem to change in individuals during treatment [80]. Beside CR, other molecules such as Fcγ receptors or adhesion molecules like ICAM-1 and LFA-1 have been suggested to contribute to FDC-mediated trapping of HIV [94].In addition to FDCs, B lymphocytes have been implicated in virus trapping and dissemination. Expression of CR1 and CR2 allows binding of immune-complexed HIV on peripheral and lymhoid B cells, which subsequently transmit the virus to T lymphocytes and promote infection [95-98]. B cells isolated from peripheral blood and LT of chronically infected individuals were shown to carry immune-complexed HIV on their surface [79]. Since B cells circulate, they potentially disseminate infectious virus throughout the body. In the LT, they pass through T cell zones containing CD4+ T lymphocytes and vice versa. In these areas, but alos in GCs, the promotion of B cell-T cell contacts sets the stage for direct transmission of trapped HIV from B to T cells (Fig. 2B). Indeed, in autologous tonsillar B cell-T cell co-cultures opsonized virus was found to preferentially bind to CR2 on B lymphocytes [98]. In this system, T cell infection occured independently of pre-stimulation of primary T lymphocytes [98]. The mechanism responsible for infection of un-stimulated primary T cells seems to involve direct interaction between B and T cells, in which B cells act as continuous supply of infectious virus. Additionally, the stabilization of B cell-T cell contacts through adhesion molecules facilitates membrane fusion between HIV and T cells [98].
It has been reported that CR3+/CR4+ dendritic cells pulsed with HIV-1 transmit the virus to freshly isolated blood monocytes and also to monocyte-derived macrophages with high efficiency [73]. Inhibition experiments using mAb against β2 integrins have demonstrated that anti-CD18 mAb significantly inhibited transmission of HIV-1 from dendritic cells to monocytes. In addition, weak inhibition of HIV-1 transmission was observed with anti-CD11b mAb, suggesting an involvement of CR3 in this process. Efficient transfer of complement-opsonized HIV was also observed from DCs to T cells in short- and long-term transmission experiments [75].
CR3 has also been detected on microglia, a functional analogue of monocytes in the brain, which play an important role in HIV neuropathology. Using an anti-CR3 monoclonal antibody, microglia can be activated and induced to proliferate via their CR3 molecules [99]. Such activated microglia cells secrete proinflammatory cytokines such as IL-1, TNF-α, IL-6 and GM-CSF. These cytokines regulate the activity of interacting cell types, induce expression of complement proteins and propagate HIV infection in the CNS [100,101].
Enhancement of HIV Infection by Anaphylatoxins
Monocytic cells of healthy subjects express high amounts of C5aR and C3aR on their surface [102,103] In contrast, studies on peripheral blood monocytes and neutrophils from AIDS patients demonstrated significantly impaired migratory activity to complement anaphylatoxins and lower expression of C5aR. As a consequence, it has been supposed that this defective migratory function may contribute to the depressed inflammatory response frequently observed in patients with AIDS [104,105].
Recently, C5a has been shown to prime monocyte-derived macrophages for HIV infection. Treatment of monocyte-derived macrophages with C5a enhanced infectivity of R5 strains up to 40-times and a similar effect was observed with a C5a derivative, C5adesArg. Kinetic analyses demonstrated that exposure to C5a accelerates productive infection in these cells. C5aR-blocking mAb reversed the susceptibility of macrophages [106] and also DCs for HIV infection [107].
Thus, increased secretion of proinflammatory cytokines correlated with higher susceptibility of macrophages to HIV infection upon cultivation in the presence of C5a and C5adesArg [106]. These observations provide a possible explanation for higher likelihood of HIV infection at the mucosal sites in individuals with sexually transmissible infections [108].