Idiopathic Inflammatory Myopathies
GENERAL PRINCIPLES
Idiopathic inflammatory myopathies are a group of heterogeneous disorders characterized by inflammation of the skeletal muscle. These can be differentiated by the pattern of muscle involvement, extramuscular manifestations, specific serologies, and findings on muscle biopsy.
Classification
• Dermatomyositis (DM): inflammatory myopathy associated with proximal muscle weakness and a characteristic skin rash. DM includes clinically amyopathic dermatomyositis (CADM) and juvenileonset DM.
• Polymyositis (PM): inflammatory myopathy that presents as proximal weakness and occasionally tenderness of the proximal musculature but lacks characteristic rash.
• Overlap syndromes: Inflammatory myopathy that occurs in the setting of another systemic rheumatic disease (i.e., SLE, systemic sclerosis, etc.).
• Anti-synthetase syndrome is a spectrum of inflammatory myopathies defined by the presence of an antibody directed against one of several aminoacyl-transfer RNA (tRNA) synthetases.
• Inclusion body myositis (IBM): inflammatory myopathy that commonly involves distal musculature and has specific muscle biopsy findings.
• Immuiie-mediated necrotizing myositis (IMNM): this inflammatory myopathy resembles PM, but muscle necrosis and minimal inflammatory infiltrate seen on muscle biopsy distinguish it from PM.
DIAGNOSIS
Clinical Presentation
• Muscle weakness: insidious onset of weakness over 3-6 months. If onset occurs in less than 2 months, suspect IMNM. All with the exception of IBM affect proximal musculature in a symmetric fashion. IBM can present with distal and asymmetric muscle involvement. A small proportion of patients can have bulbar muscle involvement. In general, muscle pain is absent or minimal.
• Cutaneous: present in DM and may precede the onset of myopathy. The most specific finding is Gottron papules, which are violaceous papules over the dorsum of MCP and PIP joints, wrists, elbows, and knees.
Other findings include a photosensitive erythematous rash on the face, scalp, anterior chest (V sign), and back and shoulders (shawl sign) and a purplish discoloration over the upper eyelids called heliotrope rash. Calcinosis is common in juvenile DM and sometimes occurs in adult DM. Patients with antisynthetase syndrome may develop hyperkeratotic, fissured skin on the palmar and lateral aspects of the fingers, called “mechanic’s hands.”• Arthritis: mild joint pain and swelling of small joints can be seen in DM and antisynthetase syndrome.
• Pulmonary: ILD is often found in DM. Other less common manifestations include diffuse alveolar hemorrhage or restrictive lung disease secondary to muscle weakness.
• Cardiac: can develop myo/pericarditis, conduction abnormalities. Heart failure is uncommon.
• Gastrointestinal: dysphagia and aspiration pneumonia can be seen in DM.
Diagnostic Testing
• Elevated muscle enzyme levels: creatine kinase (CK), aldolase, transaminases, and lactate dehydrogenase (LDH). CK can be normal or only slightly (up to 10 times upper limit of normal) elevated in IBM, moderately elevated in DM and PM and can be very high in IMNM (gt;50 times upper limit of normal).
• Autoantibodies: these are generally divided into two: myositis-specific antibodies (MSAs) and myositis-associated antibodies (MAAs). MSAs are detected primarily in patients with inflammatory myopathies and MAAs are detected in patients with other autoimmune diseases that can be associated with myositis. Different antibodies are associated with different clinical manifestations and have therapeutic and prognostic implications (Table 25-6).
MYOSITIS-SPECIFIC ANTIBODIES (MSA)
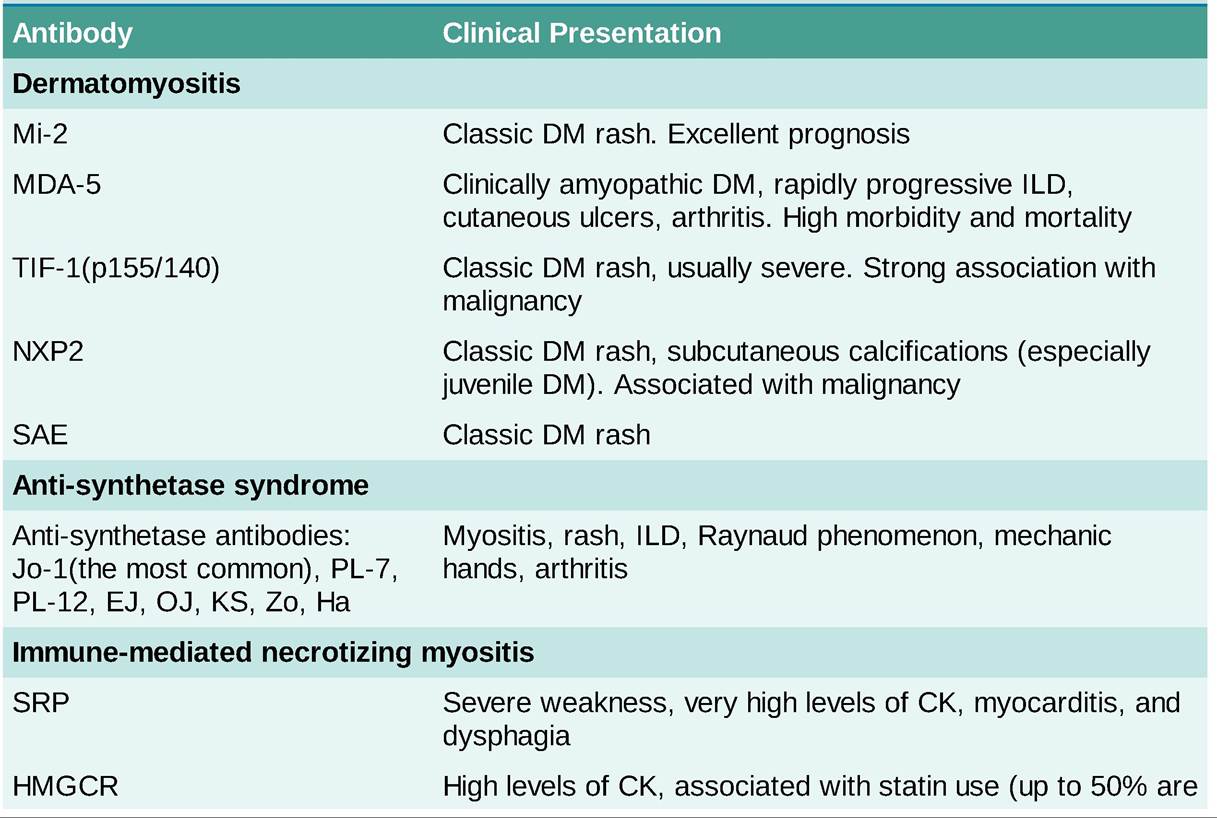

DM, dermatomyositis, IIM, idiopathic inflammatory myopathy; ILD, interstitial lung disease.
• Myositis-associated antibodies (MAAs) and their associations include the following:
î PM/Scl—overlap PM and systemic sclerosis
o U1-RNP—MCTD
î U3-snRNP (fibrillarin)—systemic sclerosis
î Ku—overlap PM and systemic sclerosis
î Ro52—frequently coupled with other MSAs. Associated with ILD
î Ro60#8725;SSA—Sjogren syndrome, SLE
o LA/SSB—Sjogren syndrome, SLE
OTHER DIAGNOSTIC PROCEDURES
• Characteristic findings can be seen on electromyogram; these include insertional activity, but these changes are not specific and can be seen in infectious or metabolic myopathies.
• A muscle biopsy can establish the diagnosis but may not be required if myositis-related antibodies are present in the right clinical setting.
• MRI is useful for the detection of muscle inflammation and necrosis and can aid in identifying a biopsy location.
• Screening for common neoplasms, such as colon, lung, breast, and prostate cancer, should be considered in these patients as well as individual risk-based assessment. Risk factors for malignancy in the setting of myositis include the presence of DM, cutaneous vasculitis, male sex, advanced age and TIF-1(p155#8725;140) or NXP-2 antibodies.
TREATMENT
• When PM or DM occurs without associated disease, it usually responds well to prednisone, 1-2 mg/kg PO daily. Systemic complaints, such as fever and malaise, respond first, followed by muscle enzymes, and finally, muscle strength. Once serum enzyme levels normalize, the prednisone dosage should be reduced slowly to maintenance levels of 10-20 mg PO daily. Appearance of steroid-induced myopathy and hypokalemia may complicate therapeutic assessment.
• Patients often need early initiation of steroid-sparing treatment and methotrexate, mycophenolate mofetil, and azathioprine are used as first-line agents.
• IV infusion of immunoglobulin are useful in patients who do not respond to steroids or have severe manifestations like truncal weakness or severe dysphagia.
• Severe cases are typically treated with rituximab, effective in patients with antisynthetase syndrome especially those that are Jo-1 positive, or intravenous immunoglobulin (IVIG).
• PM or DM associated with neoplasia tends to be less responsive to glucocorticoid therapy but may improve after removal of the malignant tumor.
• Physical therapy is essential in the management of myositis.