Charcot-Marie-Tooth (Hereditary Motor Sensory) Neuropathy
Charcot-Marie-Tooth (CMT) neuropathy (also called hereditary motor sensory neuropathy or HMSN) is a heterogenous group of inherited disease of peripheral nerve that affects both children and adults and causes significant progressive neuromuscular impairment (93,94).
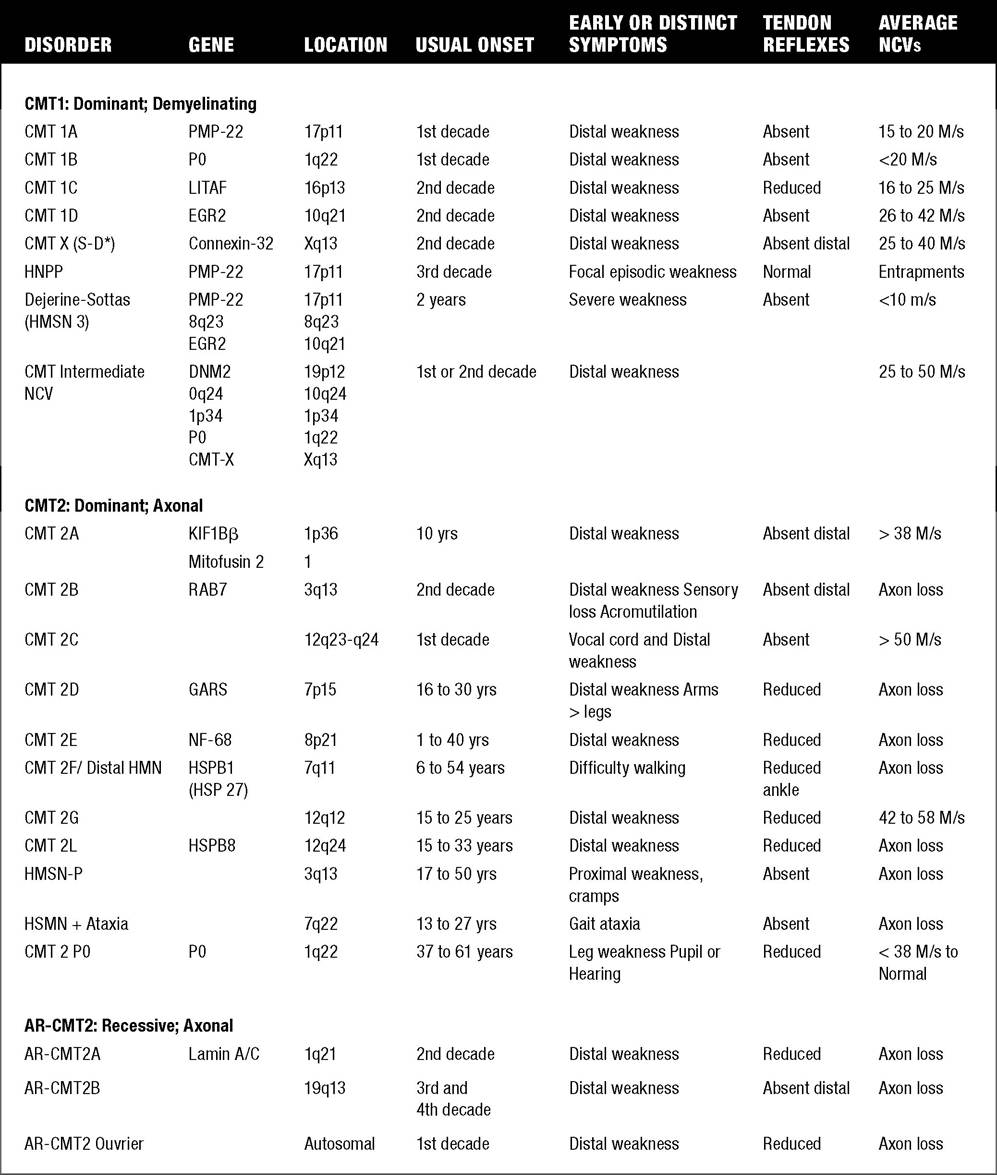
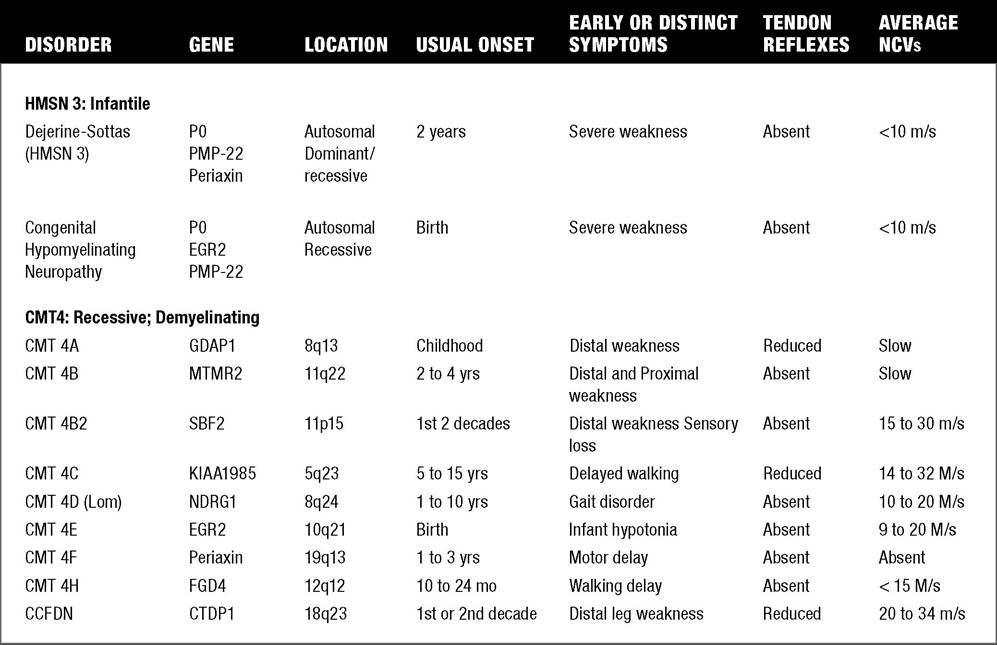
It has been estimated that 1 per 2,500 to 3,000 persons has a form of CMT. CMT 1 denotes individuals with a hypertrophic demyelinating neuropathy (“onion bulbs”) and reduced nerve conduction velocities, whereas CMT 2 refers to individuals with an axonal neuropathy and normal or slightly reduced nerve conduction velocities. Individuals with CMT 3 (Dejerine-Sotttas disease) have a primarily demyelinating peripheral neuropathy with a more severe phenotype presenting in infancy. Historically, types 1, 2, and 3 were felt to be autosomal-dominant conditions, with type 3 CMT patients exhibiting point mutations with frameshift and either dominant or recessive inheritance. CMT 4 refers to autosomal-recessive CMT. However, recently, axonal forms of CMT have been identified with autosomal recessive inheritance (deemed AR-CMT 2A, 2B, etc.)In general, in most CMT subtypes, onset is usually during the first or second decade of life. Both motor and sensory nerve function are affected. The clinical features include distal muscle weakness, impaired sensation, and absent or diminished deep tendon reflexes. Weakness usually is greatest initially present in the foot and hand intrinsics and distal lower extremities, and subsequently in the distal upper extremities. Slow progressive weakness, more proximally in the knees, elbows, and pelvic and shoulder girdles may occur over decades (56). There is variable penetrance in most subtypes. The various gene locations and known protein abnormalities associated with various forms of CMT (HMSN) are given in Table 12.3.
The majority of CMT 1 pedigrees (70%) demonstrate linkage to chromosome 17p11.2-12 and are designated CMT 1A (95).
CMT 1A duplication results in increased expression of peripheral myolin protein-22 (PMP-22). Conduction velocities are uniformly slow in all nerves, with a mean of 17-20 M/s and a range of 5-34 M/s. Onset is typically in the first decade, with leg arreflexia, gait disorder (toe-walking or steppage gait), foot muscle atrophy or pes cavus, occasionally short Achilles tendons, and enlarged nerves owing to onion bulb formation in half of patients. Distal weakness develops initially in intrinsic muscles of the feet and hands with development of wasting of musculature occurring slowly over time (Fig. 12.16). Ankle dorsiflexion, ankle eversion, and extensor hallucis longus weakness develops with more normal strength proximally. Progressive cavus foot deformities with clawing of the toes often develop (Fig. 12.17). Orthopedic procedures are limited to soft tissue procedures and correcting wedge osteotomies, and joint fusion should be avoided if possible to avoid late pain. Late in the disease, diaphragm or bulbar weakness may develop in rare cases. Progression is slow over many decades. Defects in the human myelin zero gene (P0) on chromosome 1q22-q23 leads to CMT 1B. P0 is the major protein structural component of peripheral nervous system myelin. The clinical presentation is similar to CMT1A; however, onset may lag into the second to third decade in a minority of patients and there is more variability in severity. Nerve conduction velocities are usually less than 20 m/s. P0 mutations may lead to other clinical variants, referred to as CMT 1E (demyelinating CMT with deafness), and predominantly axonal neuropathy with late adult-onset (eg, CMT 2I, and CMT 2J with hearing loss and pupillary abnormalities).CMT 2 is a less common disorder than CMT 1. Generally, CMT 2 patients demonstrate later age of onset, less involvement of the small muscles of the hands, and no palpably enlarged nerves. Wasting in the calf and anterior compartment of the leg may give rise to an “inverted champagne bottle” or “stork-leg” appearance.
Conduction velocities are mildly reduced,12.3
Hereditary Motor Sensory Neuropathy (HMSN) Types: Comparison of Clinical Features
12.3
Continued
and CMAP amplitudes and sensory nerve action potential (SNAP) amplitudes are usually reduced. CMT 2A2 with mitofusin abnormality accounts for approximately 20% of CMT 2 probands. CMT 2C linked to chromosome 12q23-q24 has interesting features of early onset in the first decade and diaphragm and intercostal weakness producing shortness of breath. Vocal cord paralysis may alter the voice of these patients. The disease may progress to proximal and facial muscles. Arthrogryposis is present in some patients. Phrenic nerve CMAPs are often reduced. CMT 2E with abnormality in neurofilament light chain (NFL) linked to chromosome 8p21 may have associated hearing loss in 30% of cases. While most axonal CMT is autosomal-dominant, emerging pedigrees are being identified with recessive inheritance.
Dejerine-Sottas disease (CMT 3) is a severe hypertrophic demyelinating polyneuropathy with onset in infancy or early childhood. Most patients achieve ambulation, but some may subsequently progress to wheelchair reliance. Nerve conduction velocities are greatly slowed (often below 10 m/s), and elevations in cerebrospinal fluid protein may be present. Dejerine- Sottas disease may be associated with point mutations in either the PMP-22, P0 or EGR2 gene (95). While this disorder was previously felt to be autosomal-recessive, many cases are due to denovo point mutations and actually have dominant inheritance.
Congenital hypomyelinating neuropathy is a severe and often fatal newborn disorder that often presents with respiratory distress in the delivery room. These infants often have severe generalized hypotonia and associated arthrogryposis.
Diagnostically, these infants have absent sensory nerve action potentials (SNAPS) or low-amplitude SNAPS with prolonged distal latencies. Compound muscle action potentials are either absent or low-amplitude, with motor conduction velocities ranging from 3-10 meters per second. The disorder has been linked to PMP-22, P0, and EGR2 genes. Sural nerve biopsy may be useful. Inheritance is usually autosomal-recessive, with some dominant inheritance linked to EGR2.Autosomal recessive CMT 4 is relatively rare. Most are demelinating with more severe phenotypes, and onset is often in childhood. CMT 4C linked to 5q23 is a relatively more common form of CMT 4.
Hereditary neuropathy with liability to pressure palsies (HNPP) is an autosomal-dominant disorder
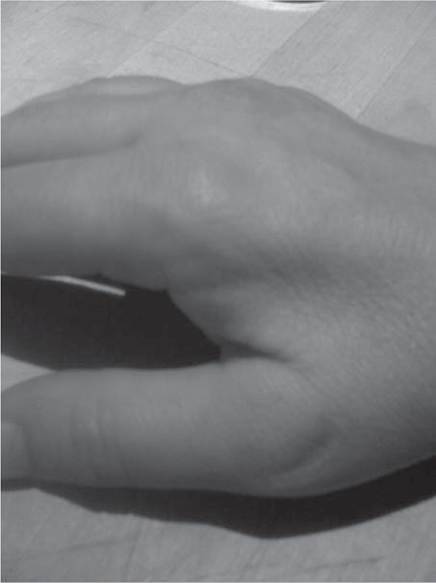
Figure 12.16 Distal weakness of intrinsic muscles of the feet (A) and hands (B) with wasting in Charcot-Marie-Tooth syndrome.
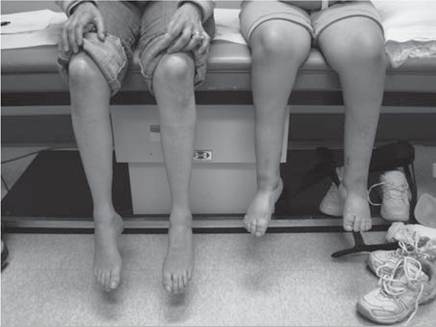
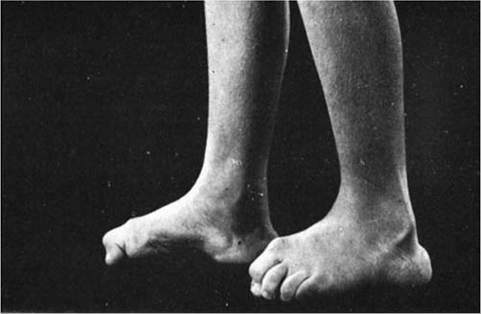
Figure 12.17 Progressive cavus foot deformities with clawing of the toes on Charcot-Marie-Tooth syndrome.
that produces episodic recurrent nerve entrapments with focal demyelination. Patients may present with peroneal palsies, carpal tunnel syndrome, and other entrapment neuropathies. A positive family history of entrapments often exists. Peripheral nerve biopsies may demonstrate segmental demyelination and tomac- ulous or “sausagelike” formations. A deletion at the PMP-22 gene locus (chromosome 17p11.2-12) causes this autosomal-dominant condition, in contrast to a duplication of this gene, which causes CMT 1A.
Patients with an X-linked dominant form of CMT (CMT-X) have been described. Male-to-male transmission is not observed, and the disorder generally shows earlier onset and faster rate of progression. The gene locus code for connexon 32 protein is Xq13, which encodes a major component of gap junctions, which provides a pathway for the transfer of ions and nutrients around and across the myelin sheath.
DNA testing for many of the CMT subtypes (particularly CMT 1) is available, but the ordering of extensive CMT batteries is expensive unless guided by nerve conduction study findings. Nerve conduction studies may be more expeditiously carried out on an affected parent to guide the molecular genetic workup of an affected child. Given the overlap of some gene abnormalities with several CMT clinical subtypes, it is often difficult to make a definitive diagnosis on genetic study results without the additional information provided by nerve conduction testing. Hereditary motor sensory neuropathy remains one clinical entity that continues to warrant electrodiagnostic evaluation.