DISORDERS OF SEX DEVELOPMENT (AMBIGUOUS GENITALIA)
Disorders of sex development (DSD) are a group of congenital conditions wherein either chromosomal, gonadal or anatomical sex development is atypical. Previous terminologies like ambiguous genitalia, intersex, pseudohermaphrodite are now avoided as they are non- informative and stigmatizing.
Physiology of sex differentiation may be divided into three components-determination of: (a) genetic or chromosomal sex, (b) gonadal sex, and (c) genital sex or phenotype (Fig. 22.4).
a. Genetic sex is determined at conception, by chance selection of the sex chromosomes in zygote—XX pair develops as potential future female and XY pair develops as potential future male.
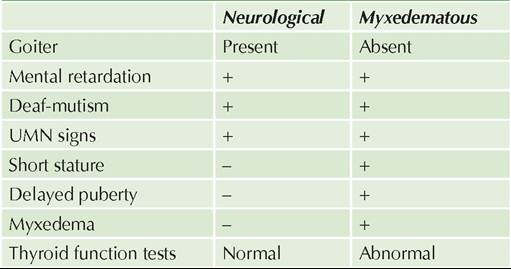
Fig. 22.4: Determination of male sex and differentiation Absence of Y chromosome, i.e. SRY gene leads to failure of above steps and development of female fetus.
*epididymis, vas deferens, seminal vesicles. **penis, scrotum, prostate DHT: Dihydrotestosterone; MIS: Mullerian-inhibiting substance; TDF: Testicular determining factor
b. Gonadal sex is determined at the end of 6th gestational week, by the presence/absence of a SRY gene on short arm of Y chromosome. Till then, fetal gonads are bipotential, capable of developing as both-the ovary or the testis. At 42nd day of gestation, the genital ridge of the XY fetus differentiates as testes in presence of SRY gene and as ovaries in its absence. Thus, presence/absence of SRY gene is one of the critical determinants of gonadal sex of fetus.
c. Internal genital sex differentiates after 8th week. Till then, both primitive sexual ducts, i.e. mullerian and Wolffian structures are present. This differentiation depends on presence/absence of male gonad, i.e. testis and not on ovaries.
In males, developing testes secrete:
• Anti-Mullerian hormone (AMH) from Sertoli cells, which leads to regression of Mullerian ducts.
• Testosterone from Leydig cells, which stimulates stabilization and differentiation of Wolffian ducts into the male internal genitals, i.e. epididymis, vas deferens, seminal vesicles.
In females, absence of testicular hormones permits development of female internal genitals with:
• Atrophy of Wolffian ducts due to the absence of testosterone, and
• Persistence of Mullerian structures in absence of AMH, to develop as upper vagina, uterus and fallopian tubes.
Development of wolffian ducts and regression of Mullerian ducts on each side depends on ipsilateral production of testosterone and MIS respectively. Hence, gonadal elements on the side determine ipsilateral internal genitalia. A streak gonad, which doesn't produce testosterone or MIS leads to development of poorly formed Mullerian structures, e.g. hypoplastic uterus on the side of streak gonad.
d. External genitals develop at 9th-13th week, depending on dihydrotestosterone (DHT)-a potent androgen, produced by conversion of testosterone in presence of 5 -reductase enzyme.
• In males, presence of DHT leads to virilization of penis and scrotum, elongation of urethra and development of prostate. Development of fetal penis is also influenced by placental human chorionic gonadotropins.
• In females, absence of testicular androgens leads to differentiation of external genitalia as females.
Types: While older classifications were based on clinical patterns, presently DSDs are classified as-(a) sex chromosomal DSD, (b) 46XY DSD, c) 46XX DSD.
a. Sex chromosomal DSD, (Previously, true hermaphrodites) denote numerical abnormalities of sex chromosomes (eg. 45X, 47XXY and mosaicisms)
TABLE 22.12 Disorders of sex development
I. Sex chromosomal DSD
• 45 X (Turner syndrome/variants)
• 47 XXY (Klinefelter syndrome variants)
• 46XX, 46XY, 45 X/46XY (Mixed gonadal dysgenesis)
II. 46 XY DSD
Disorders of testicular development:
• Complete/partial gonadal dysgenesis
• Testicular regression
• Ovotesticular DSD
Disorders of androgen production:
• Congenital adrenal hyperplasia1
• Androgen biosynthesis defects2
• Syndromic: Smith-Lemli-Opitz Syndrome
Disorders of androgen action:
• Complete/partial androgen insensitivity
Unclassified:
• Hypospadias/Epispadias
• Persistent Mullerian duct syndrome
III.
46 XX DSDDisorders of ovarian development:
• Monogenic forms
• Ovotesticular DSD
Disorders causing androgen excess:
• Congenital adrenal hyperplasia3
• Aromatase deficiency
• Iatrogenic (maternal androgen use)
• Virilizing ovarian or adrenal tumours
Unclassified:
• Mayer-Rokitansky syndrome
1STAR, 3 beta HSD, 17 alpha hydroxylase deficiencies
217 beta HSD, 5 alpha reductase deficiencies
321 hydroxylase, 11 beta hydroxylase, 3 beta HSD deficiencies
b. 46 XX DSD: (previously female pseudohermaphroditism) denote partial or complete virilization of external genitals with internal gonads as ovaries/ ovotestes with genetic sex of XX type.
c. 46 XY DSD (previously male pseudohermaphroditism) denote undervirilized or female-like external genitals with internal gonads as testes/ ovotestes and genetic sex of XY type.
Etiological classification of DSDs has been depicted in Table 22.12, though CAH and androgen insensitivity are commonest causes of 46XX DSD and 46 XY DSD respectively.
Diagnostic evaluation of these cases includes:
a. Careful family history (androgen insensitivity syndrome, CAH), previous obstetrical history of fetal, neonatal loss (salt-losing CAH) or maternal drug ingestion (estrogen/progesterone) during pregnancy.
b. External genital appearance in DSDs covers a spectrum from normal female (severe undervirilization) to genital ambiguity to normal male (severe virilization). Hyperpigmentation could be a pointer to CAH. Normal looking female genitalia with inguinal
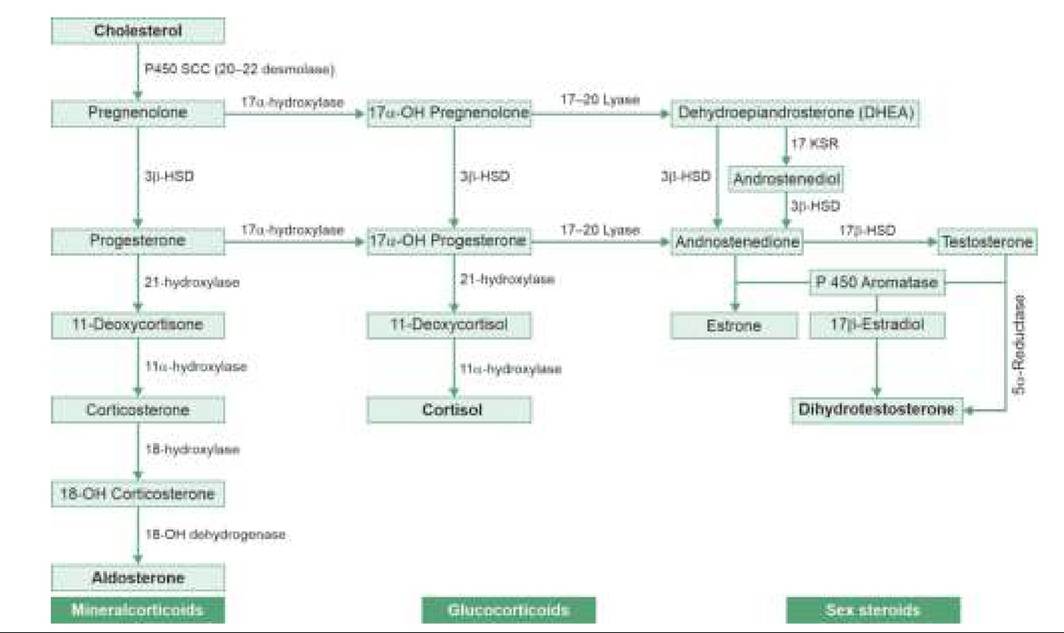
Fig. 22.5: Ambiguous genitalia.
gonads may be a case of androgen insensitivity syndrome, while normal looking male genitalia with undescended testis and empty scrotum may actually be a virilized female with CAH (Fig. 22.5).
In ambiguous appearing genitalia it is better to use neutral terms like genital tubercle (instead of penis/clitoris), labioscrotal folds (instead of labia or scrotum), gonads (instead of ovaries/ testes).
Palpation of missing testes in inguinal canal is essential to arrive at a preliminary diagnosis, before investigations.c. Investigations in all cases include:
- Determination of genetic sex by karyotyping. Rapid tests such as Y chromosome PCR for SRY gene are increasingly popular, as karyotyping may take weeks.
- Determination of internal gonads and genital structures by abdominal/pelvic sonography or laparoscopy. Very small/undescended testis may be also identified by HCG stimulation test.
- Relevant etiological investigations, dictated by clinical impression, e.g.:
#9632; Bone-age determination (advanced in CAH, delayed in aromatase deficiency),
#9632; USG/CT/MRI for central/gonadal tumors.
#9632; Chromosomal studies for Y-specific DNA sequence (Turner/Klinefelter syndrome)
#9632; Hormonal assays, e.g. testosterone/DHT ratio (5 -reductase deficiency), DHEA, cortisol and 17- OHP levels for adrenal functions, gonadotropin levels (elevated in androgen resistance)
#9632; Biochemical tests for hypoglycemia or electrolyte abnormalities (in CAH)
Gonadal biopsy is indicated only in suspected cases of gonadal dysgenesis/ ovotesticular DSD, which is very rare. Important differentiating features of various causes of DSD are summarized in Table 22.13.
TABLE 22.13: Important D/D of ambiguous genitalia
Management: Important issues in management of these cases are—(a) assignment of sex of rearing, (b) treatment of primary cause, e.g. CAH, if possible, (c) psychological support/counseling, and (d) reconstruction of external genitals and other defects, e.g. hypospadias. Removal of non-functioning gonads, especially in gonadal dysgenesis is advisable to avoid later risk of tumors.
Assignment of sex of rearing depends upon the underlying disorder and the gender identification of the individual though as a general rule:
• All 46XX should be reared as females, even if highly virilized.
• Fully/significantly feminized 46XY (such as in complete androgen insensitivity) should also be reared as females, if cause is not treatable. Estrogen replacement should be started at pubertal age for breast development in cases reared as females.
• 46XY with mild feminization or treatable cause must be reared as males.
Micropenis, a common feature in ambiguous genitalia, denotes stretched penile length of lt;2.5 cm in newborn or lt; 2SD for baby's age in older children without associated hypospadias.
Common causes of micropenis include: (a) hypothalamic- pituitary disorders, e.g. Kallmann syndrome, hypopituitarism, Prader-Willi syndrome, and (b) testicular disorders, e.g. Klinefelter syndrome, rudimentary testis or testicular regression.
In selected children, stimulation with testosterone can be used to facilitate phallic growth. Gender reassignment will be necessary in severe case or those who fail to respond to testosterone therapy.
22.6.2