Electrodiagnostic Evaluation of the Floppy Infant
The most common referral for an electrodiagnostic examination in the infant is generalized hypotonia. The most common etiology for infantile hypotonia is central, accounting for approximately 80% of cases.
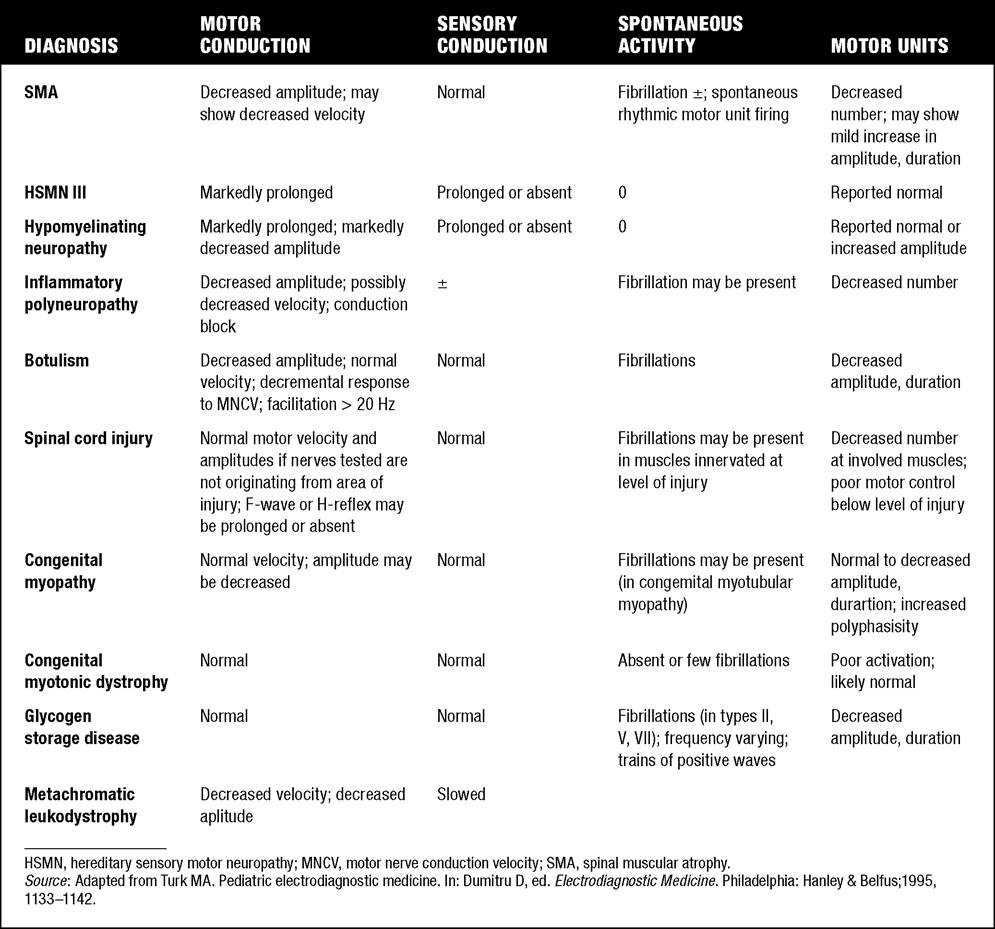
A differential diagnosis of infantile hypotonia is shown in Table 7.8 (35). Electrodiagnostic abnormalities in selected conditions producing infantile hypotonia are shown in Table 7.9.Neurogenic causes of generalized weakness in infants are more accurately diagnosed with electrodiagnostic studies than are myogenic causes (36-38). A study of the predicted value of the electrodiagnostic examination in the hypotonic infant showed that electrodiagnostic studies accurately predicted the diagnosis in 65% of infants with spinal muscular atrophy and only 10% of infants with myopathy. Seventy-five percent of the electrodiagnostic studies performed on infants with documented myopathies were considered normal (39). The sensitivity of EMG improves after age 2 (38).
In arthrogryposis multiplex congenita and hypotonia, neither muscle biopsy nor NCS/EMG alone had consistently high sensitivities, positive
7.8
Differential Diagnosis of Infantile Hypotonia
Cerebral hypotonia
Chromosome disorders
Trisomy
Prader-Willi syndrome
Static encephalopathy
Cerebral malformation
Perinatal CNS insult
Postnatal CNS insult
Peroxisomal disorders
Cerebrohepatorenal syndrome (Zellweger syndrome)
Neonatal adrenoleukodystrophy
Inborn errors of metabolism
Glycogen storage disease type II (Pompe disease)
Infantile GM1 gangliosidosis
Tay-Sachs disease (infantile GM2 gangliosidosis)
Vitamin-dependency disorders
Amino acid and organic acid disorders
Maple syrup disease
Hyperlysinemia
Nonketotic hyperglycinemia
Propionyl-CoA carboxylase deficiency
Other genetic disorders
Familial dysautonomia
Cohen syndrome
Oculocerebrorenal syndrome (Lowe)
Benign congenital hypotonia
Spinal cord
Trauma (obstetrical, postnatal)
Hypotonia early with acute paraplegia
Hypertonia
Tumor or AVM
Hypertonia may occur later or with slow-growing tumor
Anterior horn cell
Spinal muscular atrophy type I (Werdnig-Hoffman)
Spinal muscular atrophy type II
Distal SMA with vocal cord paralysis and diaphragm weakness Poliomyelitis
Neurogenic arthrogryposis
Polyneuropathies
Congenital hypomyelinating neuropathy
Chronic inflammatory demyelinating polyneuropathy
Acute inflammatory demyelinating polyradiculoneuropathy
(Guillain-Barre syndrome)
Hereditary motor-sensory neuropathies
Dejerine Sottas
Congenital hypomyelinating neuropathy
Toxic polyneuropathy
Leukodystrophies (Krabbe's, Nieman-Pick)
Leigh's syndrome
Giant axonal neuropathy
Dysmaturation neuropathy
Neuromuscular junction
Presynaptic
Infantile botulism
Hypermagnesemia—eclampsia
Aminoglycoside antibiotics
Congenital myasthenia
Choline acetyltransferase (CHAT) deficiency
Paucity of acetylcholine synaptic vesicles
Congenital Lambert-Eaton-like syndrome
Decreased quantal release
Synaptic basal lamina defects
Congenital myasthenic syndrome
Endplate acetylcholinesterase (AChE) deficiency
Postsynaptic
Neonatal (autoimmune)
Congenital myasthenia
AChR disorders involving α, β, δ, ş receptor subunits
AChR deficiency causing kinetic abnormalities in function
AChR slow-channel syndromes
AChR fast-channel syndromes
Endplate rapsyn deficiency
Myopathies
Congenital myopathies
Nemaline rod
Central core
Myotubular (centronuclear)
Mini-core (multi-core)
Congenital fiber type disproportion
Congenital myotonic dystrophy (DM1)
Congenital muscular dystrophy
Fukuyama type (CNS involvement)
Merosin deficiency (with or without CNS involvement)
Ullrich'e congenital muscular dystrophy (collagen VI deficiency, scleroatonic)
Congenital muscular dystrophy with early spine rigidity Muscle-eye-brain di sease
Walker-Warburg syndrome
Undifferentiated
Inflammatory myopathies
Infantile polymyositis
Metabolic myopathies
Acid maltase deficiency (type II)
Muscle phosphorylase deficiency (type V)
Phosphofructokinase deficiency (type VII)
Cytochrome c oxidase
Carnitine deficiency
Endocrine myopathies
Hypothyroidism
Hypoparathyroidism
AVM, arteriovenous malformation; CNS, central nervous system; SMA, spinal muscular atrophy.
7.9
Infant Hypotonia: Electrodiagnostic Abnormalities
predictive values, or specificities (40). When the clinical evaluation indicates a specific syndromic, developmental, or exogenous cause, NCS/EMG and muscle biopsy are not helpful and may not need to be performed.
When the history, examination, and genetic evaluation are unrevealing, NCS/EMG and muscle biopsy together provide valuable diagnostic information.In the evaluation of hypotonia, a complete electrodiagnostic evaluation is useful, including motor and sensory nerve conduction studies and appropriate needle examination with the highest yield muscles examined initially, and, if necessary, repetitive nerve stimulation. It should be emphasized that nerve conduction studies and electromyography are an extension of the clinician’s physical examination. Electrodiagnostic findings need to be interpreted in light of clinical examination findings. Care should be taken not to overinterpret subtle findings on needle electromyography. Low-amplitude, short-duration, polyphasic motor unit action potentials, which would be considered myopathic in adults, may be normal in young children. Motor unit amplitudes and durations may be reduced in the normal young child and mistaken for myopathic MUAPs. End-plate noise, abundant in the small intrinsic muscles of the hand and foot, may be difficult to distinguish from fibrillation potentials. Thus, borderline findings on needle EMG should not be overinterpreted in the infant and young child.
Parents should be cautioned prior to an electrodiagnostic evaluation that definitive diagnostic information is often not obtained and the results may help guide further diagnostic studies. For example, results from EMG may help to guide further studies such as muscle biopsy by providing information about the most appropriate muscle site for the biopsy. With spinal muscular atrophy, an electrodiagnostic evaluation can allow the clinician to defer a muscle biopsy and proceed with molecular genetic studies of the survival motor neuron (SMN) gene. Often, the SMN gene test is ordered prior to any electrodiagnostic studies being performed, so fewer studies have been performed on this population over the past decade. Electrodiagnostic studies in patients with hereditary motor sensory neuropathy help to categorize the neuropathy as either primarily demyelinating or axonal, and such information may help focus subsequent molecular genetic analyses. In general, nerve conduction and electromyography still provide a useful tool for the localization of lesions within the lower motor neuron, but fewer studies have been required as genetic studies have become commercially available.