HYPOTHYROIDISM
Hypothyroidism is a functional defect with decreased thyroid hormone activity, either due to deficient production or defective cell-receptors. It may be congenital or acquired and goiterous or non-goiterous.
Common causes of hypothyroidism have been enlisted in Table 22.5 while some important and common causes in childhood are as follows:Congenital hypothyroidism (CH) is the commonest preventable cause of intellectual disability in Indian children, though the incidence has declined in developed countries due to neonatal screening programs. Early diagnosis and management of congenital hypothyroidism is essential to avoid permanent growth and mental retardation, as ~70% of brain growth is over by the end of neonatal period.
CH may be transient or permanent. Transient hypothyroidism is caused by blocking of receptors by maternal antibodies or antithyroid medications in mother.
Etiology: CH may be (a) primary (~95%) due to thyroid dysgenesis, dyshormonogenesis or TSH receptor resistance, or (b) secondary (~5%) to hypothalamic or pituitary causes and very rarely, (c) peripheral due to thyroxine resistance.
Thyroid dysgenesis accounts for gt;90% cases of CH, including total thyroid aplasia (~30%), partial agenesis or ectopic thyroid tissue. Depending on the severity, these cases manifest in neonatal period or after many years.
Maternal iodine deficiency or endemic goiter, specially in sub-Himalayan region, is another important cause of CH in India, though the incidence has declined due to widespread use of Iodized salts.
Thyroxin synthesis defects are all characterized by goiterous hypothyroidism. Commonest of them being peroxidase deficiency, which prevent organification of trapped iodide into iodine.
Among the genetic factors, CH is known to be associated with TSHR, PAX8 and TSHB genes. Pendred
| TABLE 22.5: Causes of hypothyroidism | |
| Congenital | Acquired |
| • TRH deficiency/unresponsiveness | • Autoimmune thyroiditis |
| • TSH deficiency/unresponsiveness | - Hashimoto disease |
| • Thyroid dysgenesis (aplasia, ectopia) | - Polyglandular autoimmune syndromes |
| • Hypothalamic tumors (craniopharyngioma) | |
| - Iodine transport defect | • Hypopituitarism |
| - Thyroxine peroxidase deficiency | • Iodine deficiency (endemic goiter) |
| - Pendred syndrome | - Neurologic |
| - Thyroglobulin synthesis defects | - Myxedematous |
| - De-iodination defects | • Iatrogenic |
| • Intrauterine injury (maternal influences) | - Drug: Amiodarone, iodides, antithyroids |
| - Radio iodine/irradiation exposure | - Radiation exposure, radio iodine therapy |
| - Antithyroid/iodine therapy | - Thyroidectomy |
| - Endemic goiter/iodine deficiency | • Infiltrative diseases—histiocytosis |
| • TRB Antibodies (Maternal autoimmune disorders) | • Thyroid receptor non-responsiveness |
syndrome is characterized by goiter and sensorineural deafness, due to iodine-transport defect in thyroid gland and chloride-transport defect in ears.
Clinical manifestations vary according to the severity of etiological defect and most cases are asymptomatic at birth. Wide-open posterior fontanel is the only consistent, but not pathognomonic feature at birth (also seen in preterms).
Clinical features of CH may be divided into early features recognizable at birth or in neonatal period and classical clinical picture that develops only after 8-10 weeks (Table 22.6).
Typical facial features in a classic case include coarse, dull and puffy face, narrow palpebral fissure due to myxedematous eyelids, protruding tongue and depressed nasal bridge (Fig. 22.1A).
Diagnosis: Confirmation of CH depends on-a) TSH gt;20 IU/L before 2 weeks of age or gt;10 IU/L after 2 weeks, b) Low T4 lt;10 #956;g#8725;dl, or c) Low free T4 lt;1.17 ng/dl. However, TSH criteria cannot be used for diagnosis of the rare cases of central hypothyroidism due to hypothalamic/ pituitary causes, which have low TSH levels.
Neonatal screening programs use TSH estimation from the dried blood spot (DBS) sample collected after 72 hours of birth, though cord blood sample may also be used. TSH levels gt;40 IU/L in first 7 days of life, gt;20 IU/L between 7-21 days and gt;10 IU/L afterwards suggest the need to start treatment, while waiting for confirmation.
Ultrasonography and Radionuclide scan (123Tc-sodium iodide) are required to confirm thyroid agenesis, dysgenesis or ectopia (decreased uptake), which also helps to differentiate these anatomical defects form two other common cause of CH - Iodine deficiency and hormone
TABLE 22.6: Clinical features of congenital hypothyroidism
Early neonatal
• Wide-open posterior fontanel
• Prolonged jaundice
• Constipation
• Feeding/breathing difficulties
• Sluggish activity and feeble cry
• Umbilical hernia
• Hypothermia
Established case (after 8-10 weeks)
• Stunted growth (short limb-short stature)
• Typical facial appearance
• Coarse, scanty hair with low hairline
• Thick, broad neck, supraclavicular fat pad
• Broad and short hands
• Dry, thick, scaly skin with less perspiration
• Delayed motor/mental development
• Generalized hypotonia
• Delayed relaxation of deep tendon reflexes
• Delayed dentition and bone age
• Bradycardia
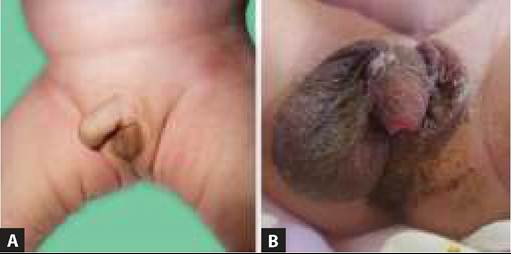
Fig. 22.1A and B: Cretinism: (A) Facial appearance; (B) absence of ossification centers at knee.
synthesis defects (normal or increased uptake). However, treatment must not be delayed for reports to avoid adverse effects on the developing brain.
CH may also be suspected on the basis of delayed skeletal maturation, i.e. absence of ossification centers for lower femur and upper tibia on knee X-ray in newborns (Fig. 22.1B) and lesser number of carpal bone centers on hand X-rays in older children. Other important radiological changes include-epiphyseal dysgenesis/ punctate epiphysis on limb X-rays, wide fontanels/ sutures and intrasutural wormian bones on skull X-rays, and deformity/beaking of T12 or L1-2.
Management: Oral sodium L-thyroxine (Eltroxin) is the drug of choice, started as 10-15 pg/kg in newborns and then the dose is titrated according to clinical response and hormone assays. T4/FT4 and TSH levels are expected to normalize by one week and four weeks respectively, though all cases should be monitored at least every 2 months in first two years. Relief of constipation is a good indicator of clinical response in most cases.
Usually, smaller doses are required in older children (4 pg/kg) in the beginning, to be titrated up over 3-4 weeks. Side-effects are dose-related and include tachycardia, headache, hyperactive behavior and features of hyperthroidism.
Life-long therapy is needed for thyroid dysgenesis or synthesis defects. In undiagnosed etiology, a therapeutic test may be attempted by stopping the drug for 2-3 weeks, after 3 years of age. Rapid rise of TSH levels after stoppage indicates need for life-long therapy.
Outcome depends on the age of diagnosis and initiation of therapy, as neonatal period is crucial for neurological growth. Without treatment, these children develop as intellectually disabled and severely short-statured. With early treatment, most cases develop well, though some may be left with borderline IQ scores, poor attention span, mild hypotonia and sensorineural hearing impairment.
Subclinical hypothyroidism with TSH lt;10 IU/L with normal FT levels is common, specially in obese children.
No treatment is required except in presence of goiter, anti-TPO antibodies or family history of hypothyroidism. However, they should be followed up cautiously and most of them revert back to normal in 3-6 months.Juvenile hypothyroidism is defined as that presenting beyond 2 years of age. It may be congenital due to late presentation of thyroid dysgenesis/dyshormonogenesis or acquired, e.g. Hashimoto's thyroiditis or iodine deficiency, discussed later.
Hashimoto's thyroiditis (Autoimmune thyroiditis), is the commonest cause of acquired hypothyroidism and the second commonest cause of goiter in children, after endemic goiter.
Etiologically it is an autoimmune disorder with presence of various antithyroid antibodies, commonest being anti-thyroperoxidase (TPO) antibodies in 90% cases or antithyroglobulin antibodies.
Positive family history is present in 25% cases and many cases are associated with: (a) chromosomal disorders, e.g. down syndrome or turner syndrome, (b) congenital rubella syndrome, and (c) autoimmune polyglandular endocrinopathies, e.g. type I (with Addison disease and mucocutaneous candidiasis) or type II (with Addison disease and insulin dependent diabetes mellitus).
Histopathologically, AIT is characterized by lymphocytic infiltration/follicle formation, followed by atrophy and fibrosis.
Clinically, most cases present in adolescent girls (male: female ratio-1:5) with two characteristic features-growth retardation and Goiter. Functionally, these cases are euthyroid (most common), hypothyroid or rarely hyperthyroid.
Clinical features depend upon duration of hypothyroidism and include firm and nodular goiter, short stature, dry scaly skin, cold intolerance, constipation, weight gain, facial puffiness, precocious or delayed puberty, menstrual irregularities, proximal muscle weakness, calf muscle pseudohypertrophy (Kocher-Debre-Semalaigne syndrome). Later course is variable and over the years, goiter may reduce or disappear with normalization of thyroid functions
Diagnosis: While definitive diagnosis requires biopsy, following supporting features are often enough make a diagnosis of Hashimoto's thyroiditis: (a) sporadic goiter in non-endemic area, (b) elevated anti-TPO antibody titers, and (c) presence of other autoimmune disorders.
Treatment depends on functional status and no therapy is required for euthyroid cases. Hypothyroid cases need Eltroxin therapy to begin with lower doses 4-6 #956;g#8725;kg and titrated up over 3-4 weeks. Eltroxin should be given on empty stomach in morning.
Follow-up hormonal assay is advised every 3 months for 2 years and then every 6 months, as the functional status may change from hypo-to hyper-thyroid state. Persistent nodular goiter despite thyroxin therapy for many months should be evaluated for thyroid carcinoma.
22.3.2