IMMUNE THROMBOCYTOPENIC PURPURA
ITP is the commonest platelet disorder in children, characterized by profound deficiency of circulating platelets due to excessive peripheral destruction (lt;1,00,000/mm3), with normal/increased number of megakaryocytes in bone marrow.
ITP may be broadly divided into:
• Primary (gt;80%), which is usually autoimmune in origin with idiopathic etiology and acute self-limiting course, and
TABLE 19.21: Causes of platelet disorders
Quantitative defects (thrombocytopenic purpura)
• Increased destruction
- Immune thrombocytopenic purpura (ITP)
#9830; Idiopathic (usually acute)
#9830; Secondary or autoimmune (usually chronic)
#9830; Neonatal alloimmune thrombocytopenia
- Mechanical
#9830; Hypersplenism
#9830; Microangiopathic hemolysis: HUS, TTP, DIC
#9830; Others: Kasabach-Merritt syndrome, drugs
• Decreased production
- BM infiltration, e.g. leukemia, histiocytosis
- Aplastic anemia
- Rare inherited disorders
#9830; Congenital amegakaryocytic thrombocytopenia
#9830; TAR syndrome (with absent/deformed radii)
#9830; Wiskott-Aldrich syndrome
Qualitative defects (platelet function defects)
• Inherited
- Glanzmann thrombocytopenia
- Thrombopathic thrombasthenia
- Gray-platelet syndrome
• Acquired
- Chronic liver/renal disease
- Drugs, e.g. aspirin, sodium valproate
HUS: Hemolytic uremic syndrome; TTP: Thrombotic-thrombocytopenic purpura; TAR: Thrombocytopenia with absent Radii
• Secondary (~20%), is usually associated with other autoimmune disorders, e.g. SLE, rheumatoid arthritis, etc., with prolonged or recurrent course.
Etiopathogenesis: Primary ITP is characterized by presence of auto-antibodies against the platelet membrane antigens, bonded to platelets.
Major antigenic target in acute ITP is PlA1 platelet antigen, i.e. glycoproteins type GPIIb/IIIa of platelet membranes.
These antibody-coated platelets are then destroyed in spleen, leading to thrombocytopenia. There is some evidence that these antibodies also act on megakaryocytes to interfere in platelet production.While exact pathogenesis for platelet-antibody formation is uncertain, acute ITP is more common in children with HLA-B8 and B12 tissue types, suggestive of a genetic predisposition.
However, history of preceding viral infection (~1-4 weeks) in gt;50% cases also suggests the role of prior sensitization for antibody production.
Clinically, primary ITP is more common in 2-8 years of age group, with equal sex distribution. Clinical signs appear only when platelet count drops below 50,000- 70,000/mm3 with acute onset of:
• Cutaneous bleeding as multiple petechial, purpuric or ecchymotic lesions, in an apparently healthy child. These lesions are earliest and often only presentation, more prominent on extensor aspects of arms and chest (Fig.
19.10). Mucus membrane bleeding, e.g. gum bleeding and epistaxis is present in ~1/3rd cases.
• Systemic bleeding like hematuria and GIT bleeding is present in severe cases (lt;20,000/mm3), though intracranial bleeding is rare till platelet counts drop lt;10,000/mm3.
• Absence of significant hepatosplenomegaly
• Self-limiting course: Acute phase lasts for ~2 weeks, when spontaneous bleeds subside though low platelet counts may persist for many weeks. Only 10-20% cases may develop chronic ITP, i.e. persistent thrombocytopenia beyond 6 months.
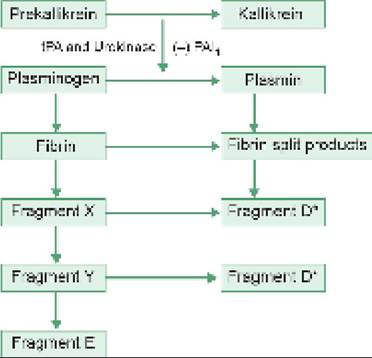
Fig. 19.10: Idiopathic thrombocytopenic purpura: (A) Purpuric spots over legs; (B) Ecchymotic patches over chest and arms.
Diagnosis is largely clinical, based on acute bleeds in an otherwise healthy child without hepatosplenomegaly, supported by:
• Prolonged BT with normal PT/PTT,
• Thrombocytopenia (lt;1,00,000/mm3),
• Presence of large platelets on peripheral smear,
• Presence of anti-platelet antibodies in gt;80% cases,
• Normal bone marrow, except increased number of megakaryocytes due to increased turnover.
Bone marrow exam is not essential for diagnosis, indicated only in cases with chronic ITP, abnormal leukocyte counts, significant anemia, gross splenomegaly and non-responsive cases.
D/D includes other causes of thrombocytopenia or bleeding disorders, e.g. (a) generalized autoimmune disorders, (b) leukemia, (c) aplastic anemia, (d) hypersplenism, and (e) platelet function defects (Table 19.22). Management: ITP is a self-limiting disease and no treatment is indicated in asymptomatic cases without wet bleeds or platelet count gt;10,000/mm3. Most guidelines recommend wait and watch policy even in cases with platelet count lt;10,000/mm3 but no wet bleeds. The aim of treatment is to prevent or treat hemorrhagic complications and not merely to restore platelet count. Management of symptomatic cases depends on the severity and includes:
• General measures, e.g. avoidance of physical trauma including intramuscular injections and drugs, e.g. aspirin. Hospitalization is indicated in cases with clinical bleeding or platelet count lt;10,000/mm3.
• Platelet transfusion is indicated only in cases with life-threatening bleeding or undergoing major surgical interventions. There is no evidence that platelet transfusions raise platelet count significantly as transfused platelets, like autologous platelets, are also destroyed by platelet antibodies.
• Immunomodulatory therapy is indicated in cases with active hemorrhage, platelet count lt;20,000/mm3, or undergoing surgical procedures. These modalities include:
- IV immunoglobulin (IVIG) therapy (1 g/kg/day for 1 or 2 days) is the treatment of choice, which prevents destruction of platelets by blocking Fc receptors of platelets and binding the circulating auto-antibodies.
TABLE 19.22: D/D bleeding disorders
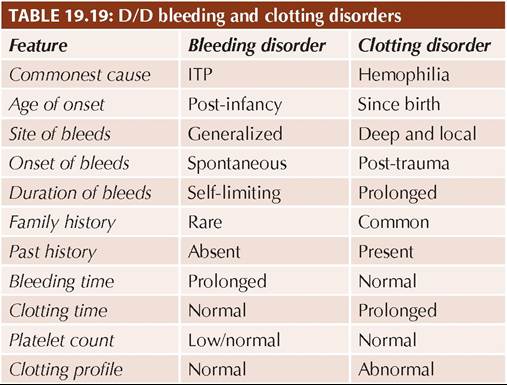
BM: Bone marrow abnormalities
- Corticosteroids, i.e. Prednisolone (PO 1-4 mg/kg/ day q6hr) until the platelet count exceed 20,000/ mm3, followed by gradual tapering.
IV methyl prednisolone (30 mg/kg/d ? 3-7 days) may be used as an alterative to IVIG therapy for rapid restoration of platelet count above safe limits.- Anti-D immunoglobulin (IV 50-75 #956;g#8725;kg) is a cost-effective alternative to IVIG, which bind the Rh+ve RBCs to produce a mild hemolysis. These antibody-coated RBCs are then preferentially cleared by spleen than antibody-coated platelets of ITP, reducing the severity of platelet destruction. Hemolysis produced by these antibodies is a therapeutic effect, rarely enough to cause significant anemia. Note that Anti-D immunoglobulins are effective only in Rh+ve children.
- Other treatment modalities like plasmapheresis (to remove platelet antibodies) or cytotoxic drugs (to prevent autoantibodies formation) are rarely needed in acute ITP.
Prognosis: Most cases of ITP resolve spontaneously in 3 weeks. However, about 20% of them may continue to have thrombocytopenia for 3-12 months (persistent ITP) or rarely even longer than 12 months (chronic or relapsing ITP).
Persistent ITP, lasting for 3-12 months, does not need specific treatment except close monitoring, aimed to keep platelet count within safe range (and not to normalize the counts) with intermittent steroids.
Chronic ITP is defined as persistent symptomatic thrombocytopenia for gt;12 months or relapse of symptoms after a few months, respectively.
These cases are more likely females and usually present with mild and insidious illness, which should be thoroughly investigated for underlying secondary causes, e.g. collagen disorder, combined variable immunodeficiency, HIV or von Willebrand disease.
For patients with chronic ITP, whose symptoms are not adequately controlled with first-line therapies or who remain dependent of corticosteroids, second-line treatment options include: (a) TPO-receptor agonists, e.g. Eltrombopag (PO 12.6-25 mg OD) or Romiplostim (1-10 #956;g#8725;kg SC once a week), (b) Other immunosuppressive agents including Rituximab, and (c) splenectomy.
As spleen is the primary site of sensitized platelet destruction, splenectomy is indicated for chronic/ recurrent ITP, refractory to medical therapy. However, it should be deferred at least till 6 years of age to reduce risk of infections. Splenectomized children are susceptible for infections by capsular organisms and should receive pneumococcal, meningococcal and H. influenzae B vaccines apart from penicillin prophylaxis.
TABLE 19.23: D/D idiopathic vs secondary ITP
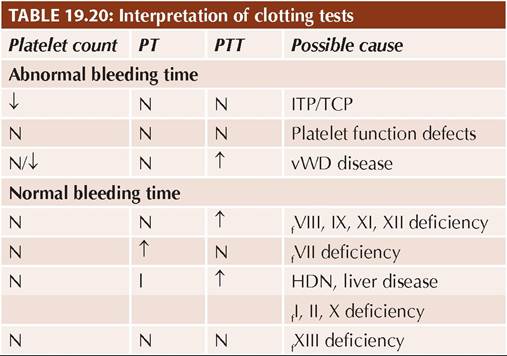
*signs of autoimmune disease, e.g. hemolytic anemia, arthritis, etc.
IV Rituximab 375 mg/ m2 weekly for 4 weeks may be used as to delay splenectomy in children though cost is prohibitive.
Some Important clinical differences between idiopathic and secondary ITP are given in Table 19.23.
Maternal ITP and Newborn
About 50% newborns of mothers with active ITP manifest with transient thrombocytopenia with/without postnatal bleeding, due to transplacental transfer of maternal platelet antibodies.
Clinical severity depends on the severity of thrombocytopenia in mother though most cases have mild disease and platelet counts recover spontaneously in next 2-4 months.
Prenatal management of these cases is limited to treatment of maternal ITP, according to her clinical status. Considering higher risk of intracranial hemorrhage during vaginal delivery, some workers advocate a single IVIG dose to mother few days before delivery followed by LSCS, though utility of these measures is controversial. Postnatal management with IVIG, steroids or platelet infusion, depends on neonatal platelet counts, till spontaneous recovery.
Other important causes of purpura in childhood include: Neonatal alloimmune Ihrobocytopenic purpura (NATP) is a rare but important cause of neonatal purpura, which may be considered as platelet equivalent of Rh-hemolytic disease. It is characterized by antigenic incompatibility between fetal platelets (PLA-1 Ag +ve) and mother (PLA-1 Ag -ve), which stimulates development of maternal antibodies.
These antibodies are then transferred transplacentally to destroy fetal platelets.Clinically, these cases may present as bleeding neonate with moderate/severe thrombocytopenia during first 2-4 weeks of life. Perinatal intracranial hemorrhage is not uncommon.
Diagnosis rests on detection of maternal alloantibodies in newborn, though prenatal diagnosis is possible in high-risk fetus (history of NATP in previous sibling) by cord-blood sampling. Unlike newborns of maternal ITP discussed earlier, maternal platelet count is normal in NATP.
D/D includes other causes of bleeding neonate specially maternal ITP and intrauterine infections (Ch 12.16.1).
Management of symptomatic cases includes repeated platelet infusions to maintain platelet counts gt;20,000/ mm3, till spontaneous recovery by 2-4 weeks.
Prevention includes—(a) genetic counseling for risk of recurrence and severity in next pregnancy (as in Rh- hemolytic disease, and (b) prenatal diagnosis in high-risk cases with fetal platelet count monitoring by repeated cordocentesis after 18 weeks. All mothers with fetal thrombocytopenia (lt;20,000/mm3) should receive weekly IVIG till term, followed by LSCS delivery (preferably) to prevent perinatal intracranial hemorrhage.
Wiskott-Aldrich syndrome, an X-linked disorder, is characterized by thrombocytopenia with tiny platelets, eczema and recurrent infections due to a molecular defect in cytoskeletal protein (WAS protein), present in platelets and T-lymphocytes.
Although thrombocytopenia may improve on splenectomy, definitive treatment requires bone marrow transplant. Lymphoreticular malignancies are common (~ 5%) in later life.
Kasabach-Merritt syndrome is a rare disorder characterized by a superficial or visceral giant hemangioma with intra-lesional platelet trapping (thrombocytopenia) and consequent activation of clotting mechanisms leading to localized DIC and hypofibrinogenemia.
Thrombotic thrombocytopenic purpura (TTP) is predominantly a disease of adults, characterized by a pentad of—(i) fever, (ii) thrombocytopenia, (iii) microangiopathic hemolytic anemia, (iv) abnormal renal functions, and (v) shifting neurological signs due to microthrombi in cerebral circulation.
An acquired deficiency of a metalloproteinase enzyme, responsible for cleaving of vWf, appears to important in etiology of TTP.
Diagnosis rests on the triad of-thrombocytopenia, hemolytic RBC morphology and abnormal renal functions, with normal coagulation studies.
Treatment is plasmapheresis, with steroids and splenectomy in resistant cases.
Platelet function defects are relatively uncommon, but should always be suspected in bleeding disorders with: (a) normal platelet count, (b) similar family history, (c) abnormal morphology/clumping of platelets on peripheral smear. A normal bleeding time does not exclude platelet function defects.
Platelet functions are commonly measured by platelet aggregometry, i.e. automated measurement of platelet clumping after addition of stimulants, e.g. collagen,
thrombin, ristocetin, etc. Some rare but important platelet function defects include:
• Glanzmann thrombasthenia is an autosomal recessive defect in platelet aggregation, due to a fibrinogen receptor (GPIIb/IIIa) deficiency in platelets. Platelet count and morphology is normal in these cases.
• Bernard-Soulier syndrome is also an aggregation defect, characterized by presence of giant platelets with thrombocytopenia, due to absence/deficiency of vWf receptors on platelet membrane.
• Gray platelet syndrome is characterized by absence of specific storage granules (#945;-granules) in platelets, causing aggregation and release dysfunction and grayish appearance of platelets on Wright staining.
19.12