NEPHROTIC SYNDROME
Nephrotic syndrome (NS) is a clinical entity related to renal disease, characterized by:
• Heavy proteinuria gt;1 gm/m2/day or 40 mg/m2/hr or UPCR gt;2 mg/mg,
• Hypoproteinemia (S.
albumin lt; 3 gm/dl), and• Presence of edema or generalized anasarca
Over 95% of NS are primary or idiopathic (INS), while the rest are secondary to an identifiable renal or systemic etiology (Table 21.9). This chapter mainly deals with INS, while important causes of secondary nephrotic syndrome are discussed in brief at the end.
Histologically, three main types of lesions are seen in INS, with significant differences in clinical course and response to therapy. Transformation from one to another histological type is uncommon.
TABLE 21.8: Causes of proteinuria
• Mild proteinuria
- Orthostatic proteinuria
- Transient: Fever, exercise, drugs*
- Non-renal: Seizures, CCF
• Tubular proteinuria
- Renal tubular acidosis
- Hereditary tubular nephropathies
- Acute interstitial nephritis
- Acute tubular necrosis
• Acute nephritic syndrome
• Nephrotic-range proteinuria
- Minimal change nephrotic syndrome (MCNS)
- Non-MCNS idiopathic nephrotic syndrome
- Secondary nephrotic syndrome
TABLE 21.9: Causes of nephrotic syndrome
Idiopathic nephrotic syndrome (INS)
• Minimal change nephrotic syndrome (85%)
• Mesangioproliferative glomerulonephritis (5%)
• Membranous nephropathy (2%)
• Focal segmental glomerulosclerosis (10%)
Secondary nephrotic syndrome
• Chronic GN: MPGN, membranous GN
• Hereditary nephropathies: Polycystic disease
• Collagen disorders: SLE, HSP
• Tumors: Leukemia, lymphoma, renal tumors
• Infections: HBV, HCV, malaria, HIV, TB, CMV
• Toxic: Insect bites, heavy metal poisoning
• Metabolic: Diabetes, amyloidosis
Congenital nephrotic syndrome
a.
Minimal-change nephrotic syndrome (MCNS) is the commonest histological type of NS in children, accounting for ~85% of cases. It is characterized by no significant abnormality on light microscopy and absence of immune-complex deposits on immunofluorescent studies. However, electron microscopy reveals extensive obliteration of epithelial foot processes. Over 95% of these cases respond well to steroid therapy.b. Mesangioproliferative nephrotic syndrome (5%) with diffuse increase in mesangial cells and matrix on light microscopy and presence of immune- complex deposits (IgM and C3) in mesangium on immunofluorescence studies. Severity of mesangial cell proliferation is related to the course of the disease. Only ~50-60% of these cases respond to steroids.
c. Focal segmental glomerulosclerosis (10%) with focal sclerotic lesions and scarring, starting from juxtamedullary glomeruli and gradually involving all glomeruli. These cases rarely respond to steroids(lt;20%) and progress rapidly to end-stage renal disease. Rare histological types of INS include Membrano-
proliferative (MPGN) or membranous nephropathy- latter being the commonest cause of nephrotic syndrome in adults (Ch 21.6.3).
Pathogenesis: Massive proteinuria is the primary pathology in NS, leading to secondary abnormalities, e.g. hypoproteinemia, edema and hyperlipidemia (Fig. 21.3). In MCNS, this proteinuria is characteristically selective, limited to loss of plasma proteins with molecular weight less than that of albumin, sparing larger proteins, e.g. globulins. On the other hand, non-selective proteinuria is common in non-MCNS variants of INS.
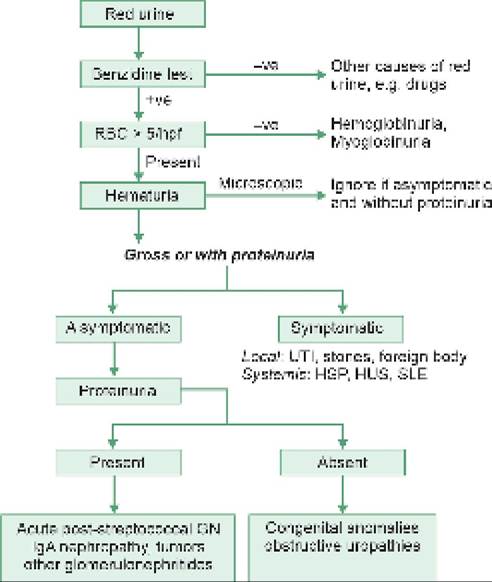
Fig. 21.3: Pathogenesis of nephrotic syndrome
LP: Lipoproteins; GBM: Glomerular basement membrane *Loss of negative charge, pore size, changes in matrix “Stimulates aldosterone/ADH mediated salt-water retention with further edema
Exact cause of proteinuria in MCNS is unclear.
Normally, plasma proteins, e.g. albumin are not filtered in glomeruli due to their size and anionic charge (presence of similar charge on GBM prevents their filtration by same-charge-repulsion phenomenon). Current evidences suggest an underlying immune dysfunction in MCNS, leading to T-cell activation and release of a cytokine-like circulating substance (yet to be isolated), which increases the glomerular permeability for albumin by interfering with the polyanionic endothelial charge or some other unknown mechanisms.Clinical presentation: MCNS commonly presents between 2 and 8 years of age, more common in males (60-70%). A preceding history of non-specific viral illness is present in some cases.
Edema is the clinical hallmark of NS, which is typically more prominent in periorbital region and during morning hours in early stages. Gradually, it progresses to a stage of generalized anasarca, with marked scrotal and abdominal wall edema. Ascites and pleural effusion may be present in severe cases. Dull abdominal pain and diarrhea is common due to mesenteric and intestinal wall edema (Fig. 21.4).
Oliguria is common in MCNS due to intravascular volume depletion, though other signs of renal disease, e.g. azotemia, hematuria and hypertension are uncommon (lt;10%) and indicate possibility of non-MCNS pathology. Diagnosis of NS depends on: (a) characteristic edema, and (b) presence of gt;2+ proteinuria on repeated testing (gt;40 mg/m2/hour or UPCR gt;2.0 mg/mg) sup-ported by,
(c) hypoproteinemia, and (d) hyperlipidemia.
Other important investigations include:
• Urinalysis for hematuria and hyaline/granular casts
• Urine culture to exclude UTI-a common precipitating event for relapses.
• Baseline Renalfunction tests, e.g. BUN, S creatinine and S electrolytes.
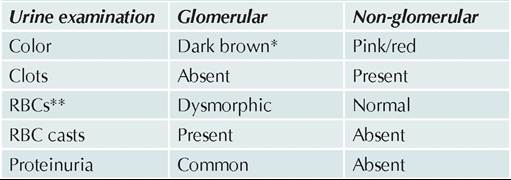
Fig. 21.4: Nephrotic syndrome: Facial puffiness, ascites and edema, specially scrotal edema (Inset)
(Courtesy: Dr C T Deshmukh)
• S Complement (C3) levels, which are essentially normal in MCNS, but may be reduced in non-MCNS variants or APSGN.
• Investigations to exclude underlying infections, e.g. chest skiagram, TT and HIV/HBV serology, before starting steroid therapy.
Renal biopsy is not routinely indicated except in cases of probable atypical nephritic syndrome (non-MCNS pathology), as indicated by—(a) age of onset lt; 1 year or gt; 12 years, (b) gross hematuria or persistent hypertension/ azotemia, (c) persistently low C3/C4 levels, (d) no response to steroid therapy after 8 weeks, and (e) features of underlying connective tissue disorders, e.g. SLE or IgA vasculitis, chronic infections, e.g. hepatitis B or C and syndromic features, e.g. Nail-Patella syndrome.
D/D of NS includes:
• Other causes of proteinuria/edema — commonest being APSGN (Table 21.8).
• Differentiation between MCNS and non-MCNS disease (Table 21.10).
Course: Over 90% of initial attacks respond well to steroid therapy, though subsequent course is variable and often unpredictable, interrupted by recurrent relapses. Relapses are more likely in younger cases and within first few weeks of remission. Most steroidresponsive MCNS cases stop getting relapses by late adolescence, without residual renal damage. However, steroid-resistant cases with significant renal lesions (non- MCNS) usually progress to ESRF.
Common terms, used to denote course of NS are:
• Remission: Urine albumin nil or trace (lt;4 mg/m2/ hr or UPCR lt;0.2 mg/mg or negative dipstick) for 3 consecutive days;
• Relapse: Reappearance of proteinuria (gt;2+ or gt;40 mg/ m2/hr or UPCR gt;0.2 mg/mg) for 3 consecutive days, having been in remission previously;
• Frequent-relapser: Two or more relapses in first 6 months or three or more relapses in any 12 months;
TABLE 21.10: D/D MCNS vs. non-MCNS
| MCNS | Non-MCNS | |
| Age | 2-8 years | gt; 8 years |
| Sex | Mlt;F | Equal |
| Hematuria | lt;10% | Common |
| Hypertension | lt;10% | Common |
| Azotemia | Rare | Common |
| Selectivity* | Yes | No |
| Serum C3 | Normal | 4 |
| Steroid response | Excellent | Poor |
| Biopsy | MCNS | Diagnostic |
| *of proteinuria | ||
• Steroid-dependent: Two consecutive relapses when on alternate day steroids or within 14 days of its discontinuation;
• Steroid-resistant: Absence of remission, despite 6 weeks of daily prednisolone (2 mg/kg/day) therapy;
• Complicated relapse: Relapse with complications requiring hospitalization, e.g.: (a) severe hypovolemia, (b) severe edema, (c) severe infection, e.g.
peritonitis, cellulitis or meningitis, or (d) thrombosis;• Significant steroid toxicity with hyperglycemia, obesity, short stature, raised intraocular pressure; cataract, myopathy; osteonecrosis or psychosis.
• Difficult to treat steroid-sensitive disease refers to- (a) frequent relapses or significant steroid toxicity with infrequent relapses, and (b) failure of two or more steroid-sparing agents (including levamisole, cyclophosphamide, mycophenolate mofetil);
Complications are more common during relapses than in initial attack and include:
• Infections due to edematous skin, impaired phago- cytic/opsonin activity and immunosuppressive therapy. Signs of infections and inflammation are often masked in NS, specially in cases on steroid therapy, and diagnosis depends on high risk of suspicion as well as relevant investigations.
Spontaneous bacterial peritonitis (SBP) is the most important infective complication in NS, which should be suspected in any case with persistent abdominal pain and may need exclusion by abdominocentesis. Other important infections include cellulitis, bone and joint infections, pneumonia, meningitis, UTI, etc.
Infections due to capsulated organisms, e.g. pneumococci, Salmonella and H. influenzae, are most common in NS, due to urinary loss of B-factor and consequent impairment in alternate complement pathway. Immunization with influenza, pneumococcal and varicella vaccine is advisable in all nephrotic children.
• Thromboembolic complications are common due to a hypercoagulable state in NS following hemoconcentration due to hypovolemia, elevated fibrinogen levels, and urinary loss of anticoagulants, e.g. antithrombin III, protein C and protein S. These cases may present with renal vein thrombosis, cerebral strokes or pulmonary embolism.
• Hypovolemic shock, may develop due to extravascular movement of fluids in hypoproteinemic state, further complicated by diuretics. These cases may develop pre-renal failure with urinary sodium is lt;10 mEq/L).
Intrinsic renal failure is rare in MCNS.• Edema-related complications, e.g. skin cracks, sores and infections, are common in NS with severe edema.
• Iatrogenic complications secondary to long-term steroid therapy, present as steroid facies (moon face with hirsutism), obesity, growth failure, hypertension, cataracts, glaucoma, hyperglycemia, osteoporosis and avascular necrosis. Immunosuppression may lead to reactivation of tuberculosis and/or severe presentation of common illnesses, e.g. measles and varicella.
Management aims for—(a) control of proteinuria, (b) prevention, early diagnosis and treatment of relapses, and (c) management of complications. Hospitalization is necessary during initial attack for confirmation of diagnosis and in complicated relapses, while uncomplicated relapses may be treated at home. Important steps in management (Fig. 21.5) are as follows:
I. Management of first attack involves:
• Control of edema: Generalized edema is the most important presenting manifestation of initial attack/ relapse, which should be managed according to its severity, as follows:
- No anti-edema measures are necessary in mild edema except salt restriction (lt;2 mEq/kg/d), as steroid therapy itself induces diuresis within one week. Usual amount of salt in diet is permitted but salty snacks or additional intake should be avoided.
- Moderate edema needs some salt-restriction along with diuretics, e.g. PO/IV Furosemide (2 mg/ kg/d q8-12hr). Furosemide is a potent diuretic and can cause sudden hypovolemia and electrolyte disturbances, specially Hypokalemia. Simultaneous administration of oral potassium supplements (PO1-2 mEq/kg/day) or a potassium-sparing diuretic, e.g., spirolactone (PO 1-3 mg/kg/day) is advisable.
- In severe edema with marked hypoalbuminemia (Serum Albumin lt;1 gm/dl), diuretics alone are ineffective and hazardous due to intravascular volume contraction. These cases should receive an IV infusion of human salt-free albumin 5% (0.5-1 gm/kg over 60-90 minutes) to bring back the extravascular fluid in intravascular compartment, before Furosemide therapy.
Other supportive measures in severe edema include salt and fluid restriction, mechanical measures, e.g. scrotal support to reduce scrotal edema, and abdominocentesis in cases with respiratory embarrassment due to severe ascites.
• Exclusion of co-existing infections, e.g., UTI, tuberculosis, etc. is essential before initiating steroid therapy and requires thorough clinical examination as well as baseline investigations, e.g. urine culture, chest skiagram and tuberculin test in all cases.
• Control of proteinuria: Immunosuppressive therapy with steroids is the cornerstone of the treatment in INS, irrespective of histopathology. However, it is preferable to delay it for few days after hospitalization to: (a) reduce severe edema as steroids may cause further fluid retention, (b) exclude underlying infection, e.g. UTI or tuberculosis, and (c) wait for spontaneous remission, which is possible in some cases.
Prednisolone is preferred over other steroids due to relatively less salt/water retention and effect on endogenous cortisol levels. While various regimens are used, Indian Society of Pediatric Nephrology (ISPN) recommends treatment of the first attack with Prednisolone, as follows:
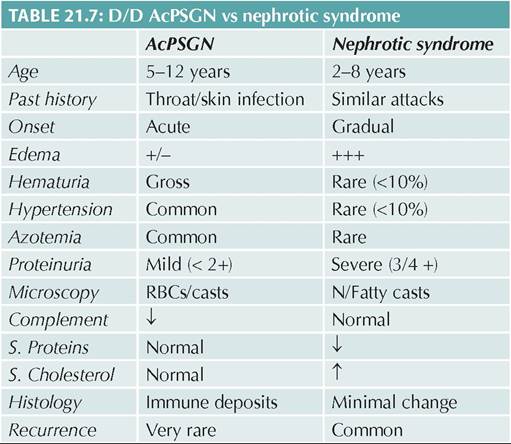
*See text, CNI: Calcineurin inhibitors
Fig. 21.5: Managemant algorithm for nephrotic syndrome
21
- PO 60 mg#8725;m2#8725;day or 2 mg/kg/day (maximum 60 mg) in one/two divided doses for 6 weeks, followed by
- PO 40 mg/m2/day or 1.5 mg/kg/day (maximum 40 mg) as a single morning dose on alternate days for next 6 weeks.
Single morning dose is preferred in later half of regimen to minimize the effect of exogenous steroids on normal diurnal rhythm in cortisol secretion. Therapy based on body surface area is preferred in young children. After completion of regimen, steroids may be stopped abruptly, without tapering. While most cases achieve remission within 10-14 days, therapy should continue for 12 weeks irrespective of the day of response, to reduce the risk of early relapse.
• Regular monitoring with daily weight record, intakeoutput chart and urine examination for proteinuria, till discharge. On discharge, parents must be trained to do daily urine examination for early detection of relapse.
• Treatment of complications:, e.g. infections, thromboembolic episodes, hypertension, etc. However, prophylactic antibiotics, antihypertensives or anticoagulants are not recommended.
II. Management of subsequent relapses: Intermittent low-grade proteinuria (gt;2+) is often triggered by minor infections and disappears spontaneously after control of infections. Persistent proteinuria (gt;2+) for 3 consecutive days indicates relapse and must be treated on same lines as for first attack but with some differences in duration of prednisolone therapy, as follows:
• PO 60 mg/m2/day or 2 mg/kg/day in one/two divided doses, until urine protein is trace or nil for 3 consecutive days; followed by
• PO 40 mg/m2/day or 1.5 mg/kg/day as a single morning dose on alternate days for next 4 weeks.
III. Management of frequent-relapsers or Steroiddependent cases aims to achieve and maintain remission while minimizing the side-effects of steroids, using long term low-dose steroids along with steroid-dose sparing agents.
After the initial treatment of a relapse as described earlier in “management of subsequent relapsesquot;, these cases should be treated with:
a. Gradual tapering of the steroid dose to minimum possible, required to keep the patient albumin-free (0.5-0.7 mg/kg alternate days) for 9-12 months;
b. Switch-over of same dose on daily basis (rather than alternate day) for 5-7 days during fever or respiratory infections
c. Addition of a steroid-sparing agent in patients failing on alternate-day steroids, steroid toxicity or complicated relapses as follows:
- PO Levamisole (2-2.5 mg/kg as single dose alternate day) for 1-2 years. Alternate-day steroids may be tapered and discontinued after 3-4 months of starting levamisole. Leukopenia is the main side-effect of Levamisole therapy, which should be monitored every 2 months.
- PO Mycophenolate mofetil or MMF (PO 600-1200 mg/m2/day in two divided doses) for 12-36 months along with tapering doses of steroids during initial 3-6 months. Gastrointestinal upset and leukopenia are main side-effects. Therapeutic drug monitoring (TDM) is recommended, if possible. Therapy should be started with lower range of the dose and increased gradually, if TDM is not possible.
- PO Cyclophosphamide (PO 2-2.5 mg/kg/daily for 8-12 weeks) may induce long-lasting remission in 30-40%, when given along with alternate day Prednisolone (1 mg/kg/day). Hemorrhagic cystitis is common and may be prevented with adequate hydration. Long-term gonadal toxicity and risk of malignancy precludes a second course of treatment.
- PO Calcineurin inhibitors, e.g. Cyclosporine (4-5 mg/kg/day in divided doses) or Tacrolimus (0.1-0.2 mg/kg/d in divided doses), are necessary for 12-24 months in cases with difficult-to-treat steroid sensitive NS after failure of 2 of abovementioned steroid-sparing agents. Renal biopsy is indicated before starting calcineurin inhibitors and TDM is recommended to adjust the doses for both drugs. Important side-effects of Cyclosporine include hirsutism, hypertension and hyperlipidemia, while tacrolimus leads to hyperglycemia, tremors and seizures. Both drugs are potentially nephrotoxic and renal functions, lipid profile and blood sugar should be monitored every 3 months.
- Rituximab-a monoclonal anti-CD20 antibody (IV 375 mg/m2 slow infusion) may be used in steroid-dependent NS, who do not respond to other steroid-sparing agents. Considering severe immunosuppression and acute allergic reactions as important side-effects, Patient should be screened for HIV, HBV and HCV prior to the infusion and monitored for acute lung injury or anaphylaxis/ serum sickness during or soon after infusion.
IV. Treatment of steroid-resistant NS: Steroid-resistant NS includes those who do not respond during initial attack (primary resistance) or during relapse (secondary resistance). All these cases require:
• Renal biopsy before further treatment, which usually reveals FSGS or MPGN histology,
• Genetic studies in cases of—(a) congenital NS, (b) primary steroid resistance with onset in infancy, (c) familial steroid-resistance, (d) syndromic NS, and (e) primary steroid-resistance not responding to even calcineurin inhibitors; as many of them have mutations in NPHS2, NPHS1, WT1 and other genes.
• Screening for secondary causes of NS, e.g. connective tissue disorders.
While many regimens are used in these cases, therapeutic response is generally unsatisfactory, specially in those with non-MCNS histology or monogenic disease.
Suggested therapeutic modalities, irrespective of histology, include a combination of calcineurin inhibitor (Cyclosporin/Tacrolimus) along with an ACE inhibitor (Enalapril/ Ramipril) and tapering doses of Prednisolone. Patients who respond, usually respond within 3-6 months and even partial response (1+ or 2 + proteinuria or UPCR 0.2-2.0 mg/mg) is acceptable in them.
Patients with monogenic disease are unlikely to respond to immunosuppression and should be maintained on ACE inhibitors and supportive therapy.
ACE inhibitors reduce severity of proteinuria by altering the capillary permeability and reducing glomerular hydrostatic pressure. Side-effects include persistent dry cough or hyperkalemia. Angiotensin receptors blockers, e.g. losartan may be used in cases with persistent cough on ACE inhibitors. Rituximab may be tried in cases after excluding monogenic disease.
Severe hypercholesterolemia may be treated with HMG-coenzyme-A reductase inhibitors.
However, ultimate prognosis is poor in steroid- resistant NS and most cases progress to ESRD in second decade of life. Renal transplant is the last effective option in them, though glomerular lesions tend to recur in transplanted kidney in 15-55% cases.
Other important causes of nephrotic syndrome are as follows:
Congenital nephrotic syndrome is defined as the presence of nephrotic syndrome within 3 months of age. Most of these cases have classic 'Finnish type' disease, though many other conditions are responsible for small number of cases (Table 21.11). Renal biopsy is essential in all cases of congenital NS and response to medical treatment is unsatisfactory, though some patients benefit from renal transplantation.
Finnish-type NS is an autosomal recessive disorder (NPHS 1,2 gene), characterized by typical renal biopsy, i.e. mesangial sclerosis and microcystic dilatation of proximal convoluted tubules. These cases are usually born prematurely with very large placenta and present in neonatal period with progressive edema, failure to thrive,
TABLE 21.11: Causes of congenital nephrotic syndrome
• Finnish type
• Diffuse mesangial sclerosis
- Denys-Drash syndrome
- Galloway-Mowat syndrome
• Primary focal segmental glomerulosclerosis
• Intrauterine infections: TORCH, syphilis, HBV recurrent infections, thromboembolic complications and progression to renal failure within 2-3 years. Elevated alpha-fetoproteins in mother and amniotic fluid suggest possibility of Finnish NS.
Treatment is symptomatic and supportive with periodic IV albumin infusions (every 2-3 weeks), judicious use of diuretics, nutritional supplements and treatment of complications. These cases are steroid- resistant and rapidly progress to ESRD, though ACE inhibitors or Indomethacin may reduce proteinuria in some cases. Bilateral nephrectomy with renal transplant is helpful in many cases.
Secondary nephrotic syndrome is rare in childhood and usually related to inflammatory or coagulatory glomerular insult (Table 21.9). Some important glomerulopathies presenting as nephrotic syndrome have been discussed in Ch 21.6.
21.8