Neurocutaneous syndromes
Neurocutaneous syndromes (phakomatoses) include a heterogeneous group of disorders characterized by abnormality of both integuments-skin and central nervous system. Most of these disorders are familial (except Sturge- Weber syndrome), believed to arise from a defect in differentiation of the primitive ectoderm.
Important neurocutaneous syndromes include neurofibromatosis, tuberous sclerosis, Sturge-weber disease, Ataxia-telangiectasia, Von-Hippel Lindau disease, Linear nevus syndrome, Hypomelanosis of Ito and Incontinentia pigmenti, of whom the first four are relatively common and discussed here.
Neurofibromatosis (NF) is the commonest neuro- cutaneous disorder that represents an abnormality of neural-crest differentiation and migration with two distinct types-more prevalent NF1 or Von Recklinghausen disease, with an estimated incidence of 1:4000, and a rare variant NF2, contributing ~10% cases.
Etiology: NF is an autosomal dominant disease though half of the cases are due to spontaneous mutations. Deletion or inactivation of NF gene on chromosome 17 is responsible for NF1, while NF2 gene is probably located at chromosome 22.
Clinical manifestations: NF is a progressive neuro- cutaneous disorders with development of features and complications even beyond childhood. Important clinical indicators of NF include:
a. Oculo-Cutaneous manifestations, e.g.
• Cafe-au-lait spots, i.e. hyperpigmented oval macules over trunk and extremities, sparing the face usually appear in infancy and increase in number and size over time. While also present in normal children,
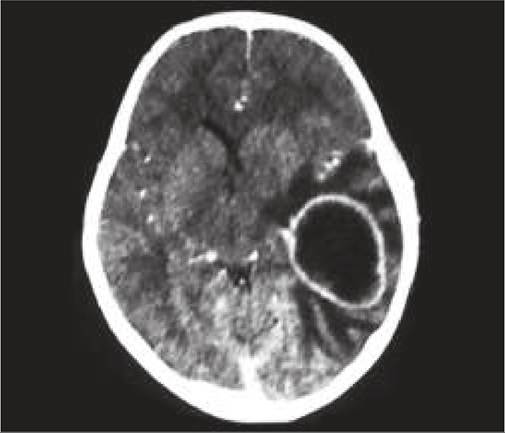
Fig. 18.17A: Neurofibromatosis: Cafe-au-lait spots.
Fig.
18.17B: Neurofibromatosis: Neurofibroma.Six or more Cafe-au-lait spots of gt;5 mm are hallmark of NF (Figs. 18.17A and B).
• Axillary or Inguinal freckling, i.e. multiple hyperpigmented lesions of gt;2-3 mm in size.
• Lisch Nodules, i.e. small-pigmented hamartomas on the iris. Posterior subcapsular lenticular opacities are common in NF2.
• Neurofibromas, i.e. small, rubbery, purplish nodular lesions over skin arising from Schwann cells or nerve sheaths. These lesions usually appear or enlarge during adolescence, though Plexiform lesions, i.e. diffuse thickening of nerve trunks in orbital or temporal region may be present at birth.
In addition, orthopedic complications, e.g. scoliosis, sphenoidal dysplasia and cortical thinning of long bones, etc. are common in older children.
b. Neurological manifestations are more common in NF1, though may not be present at the time of diagnosis and include seizures, learning disabilities, speech defects, hydrocephalus/macrocephaly and transient ischemic attacks with hemiparesis.
c. Other neoplastic lesions, e.g. optic glioma, acoustic glioma, meningioma, pheochromocytoma, leukaemia and Wilms tumor. Optic gliomas with/without vision defects are more common in NF1, while bilateral acoustic neuromas are distinctive features of NF2.
Diagnosis is largely clinical, as follows:
• Diagnostic criteria for NF1 (2021) include presence of any two of the following (or any one feature with NF1 in either parent):
- Six or more Cafe-au-lait spots gt;5 mm in greatest diameter,
- Axillary or inguinal freckling,
- Two or more Lisch Nodules or choroid abnormalities
- Two or more Neurofibromas of any type or one plexiform neurofibroma,
- Distinctive osseous lesions, e.g. sphenoid dysplasia, cortical thinning of long bones, etc.
- Optic nerve glioma with/without vision defects,
- Heterozygous pathogenic variant in gt;50% of normal tissue, e.g. WBCs.
• Diagnostic criteria for NF2 include:
- Bilateral acoustic neuroma on CT/MRI or
- Family history of NF2 in first-degree relatives with:
#9632; Unilateral acoustic neuroma, or
#9632; Any two of the following—(i) neurofibroma, meningioma, (iii) glioma, (iv) Schwannoma or (v) posterior subcapsular lenticular opacities.
No investigations are needed except neuroimaging and EEG in cases with neurological features.
Prenatal diagnosis by fetal DNA analysis is indicated in cases with family history.
Treatment is non-specific, except follow-up for detection of complications and tumors. Neurofibromas of peripheral nerves are indolent and largely cosmetic lesions and need not be removed unless compressing the nerves, or subjected to repeated irritation/trauma or develop malignant changes. Some plexiform neuromas may be removed for cosmetic purpose.
Tuberous sclerosis (TS) is a heterogeneous disease with wide clinical spectrum affecting many organs, e.g. skin, brain, heart, kidney, eyes, lungs and bone.
Etiologically, it is inherited as autosomal dominant disease (with two identified gene loci-TSC1 at chr9q34, and TSC2 at chr16p13), though ~ 50% cases are sporadic mutations. TSC1 gene encodes a protein hamartin, while TSC2 encodes another protein tuberin.
Pathologically, typical brain lesions consist of subependymal tubers, which undergo calcification and project into the ventricular cavity, producing candle-dripping appearance.
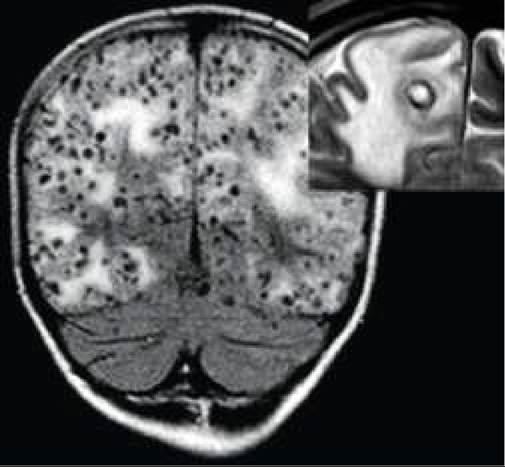
Fig. 18.18A: Tuberous sclerosis: Adenoma sebaceum.
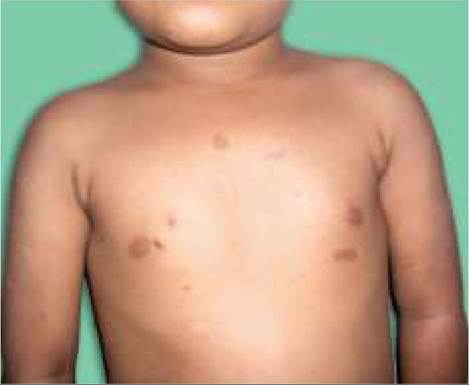
Fig. 18.18B: Tuberous sclerosis: Dripping candle appearance on CT scan.
Clinical spectrum is extremely heterogeneous, ranging from asymptomatic disease except few cutaneous markers to severe intellectual disability and refractory seizures in same family.
Intractable infantile spasms may be earliest indicator of TS in first year of life, though most cases present beyond infancy with:
• Cutaneous lesions:
- Ash-leaf macules, i.e. characteristic hypopigmented lesions over trunk and limbs are earliest cutaneous markers, present since birth in 90% cases and best visualized by Wood's ultraviolet lamp.
- Adenoma Sebaceum, i.e. tiny red or pink nodules over face mainly in perinasal region, generally seen from 4-6 years onwards, which may coalesce later to give a fleshy appearance (Fig. 18.18A).
- Shagreen Patch, i.e. rough, raised lesions with orange peel consistency, usually in lumbosacral region.
- Periungual fibromas with fleshy nodules around nails, developing during adolescence.
• Neurological manifestations:
- Recurrent seizures, as infantile spasms in infancy or generalized seizures in later childhood.
- Intellectual disability, more severe in cases with early presentation and infantile spasms.
- Hydrocephalus, due to obstruction of CSF flow at foramen of Monro by tubers.
- Presence of calcified tubers in periventricular area, from 3-4 years onwards on MRI.
- Retinal Lesions, e.g. mulberry tumors or phakoma (flat-gray lesions) over or around optic disc.
• Extra-CNS tumors, e.g. rhabdomyoma of the heart, renal tumors and angiomyolipomas in lung. Rhabdomyomas are commonest tumors in TS, seen in ~50% cases. While these tumors may be detected in utero on fetal echocardiography and lead to CCF and arrhythmia in infancy, most of them resolve spontaneously with age.
Diagnosis: Apart from cutaneous markers, confirmation of TS requires demonstration of one or more calcified periventricular tubers on CT/MRI, often giving a Dripping Candle appearance (Fig. 18.18B).
Since calcified tubers may not be visible on CT scan before 3-4 year of age, MRI is more useful for early detection of uncalcified tubers and alterations in cortical cyto-architectonics. EEG may reveal hypsarrhythmia in cases with infantile spasms.
Diagnostic criteria of TS include presence of any two of eleven major criteria or any one major and two of the six minor criteria.
• Major criteria include-Hypomelanotic macules gt;3-5 mm. Three or more angiofibroma, Two or more Ungual fibroma, Shagreen patch, Multiple renal hamartoma, Cortical dysplasia, Subependymal nodules, Subependymal giant cell astrocytoma, Cardiac rhabdomyoma, Lymphangioleiomyomatosis and two or more Angiomyolipoma.
• Minor criteria include Confetti skin lesions, three or more dental enamel pits, two or more intraoral fibroma, retinal achromic patch, multiple renal cysts and non-renal hamartomas.
Treatment of TS includes—(a) control of seizures by anti-epileptic drugs, (b) management of hydrocephalus with shunt surgery, (c) follow-up for early detection of renal, cardiac or pulmonary lesions and (d) genetic counseling. TS associated Infantile spasms respond better to vigabatrin than ACTH.
Everolimus (MTOR inhibitor) has been approved from TS with refractory epilepsy, subependymal giant cell astrocytoma and renal angiomyolipoma.
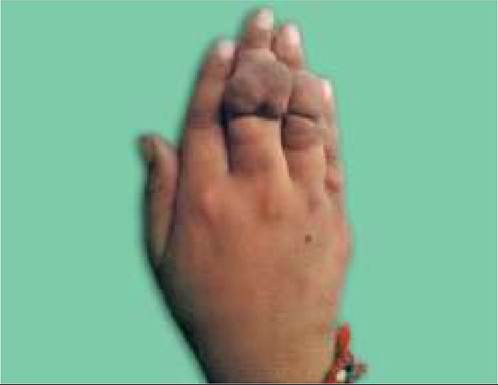
Fig. 18.19A: Sturge-Weber syndrome: Facial hemangioma.
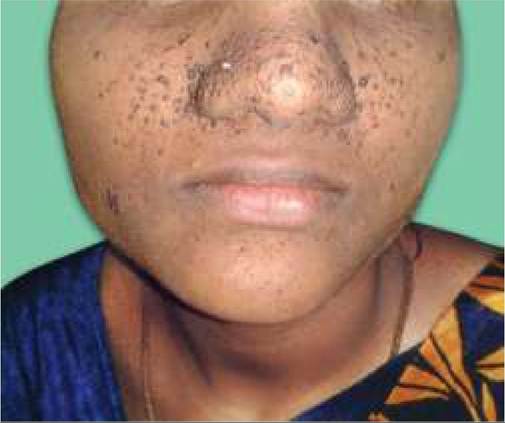
Fig. 18.19B: Sturge-Weber syndrome: Tram-track appearance.
Rarely, subependymal nodules may increase in size and turn malignant and periodic MRI every 2-3 years is recommended to monitor size of these nodules.
Sturge-Weber syndrome (SWS) is characterized by a combination of facial and intracranial hemangiomatosis, presenting with recurrent seizures, focal motor deficits and intracranial calcifications.
Etiology: SWS is not hereditary, but related to somatic mutations in GNAQ gene. It is thought to result from anomalous development of primordial cephalic vascular bed during early stages of vascularization.
Clinical features include:
• Facial nevus (Port-wine stain) since birth, mainly over upper face and eyelid (Fig. 18.19A).
• Neurological features, e.g. contralateral focal seizures due to intracranial hemangioma, with/without progressive hemiparesis, mental retardation or learning disabilities.
• Ocular features, e.g. Buphthalmos and glaucoma in ipsilateral eye.
On Roach scale, SWS is classified as—(a) Type I with facial and intracranial angiomas, (b) Type II with Facial angioma without CNS involvement and (c) Type III with Intracranial angiomas without facial involvement.
Diagnosis depends on combination of facial and neurological features and presence of typical intracranial calcifications on skull X-rays termed as Rail-road or Tram-track appearance (Fig 18.19B). CT scan may reveal calcification and ipsilateral cortical atrophy with ventricular dilatation.
Treatment includes—(a) control of seizures, (b) surgical lobectomy/hemispherectomy in refractory seizures, (c) treatment of facial nevus with laser therapy, and (d) regular monitoring of intraocular tension for glaucoma.
Ataxia telangiectasia, an autosomal recessive disorder due to mutation in ATM gene on chromosome 11q, is characterized with slowly progressive cerebellar ataxia from 1-3 years of age and telangiectatic vascular lesions over skin and conjunctiva. Choreoathetosis and ocual apraxia is common.
Common associated abnormalities include IgA deficiency leading to recurrent sinopulmonary infections and lymphoreticular malignancies.
18.15