URINARY TRACT MALFORMATIONS
Urinary tract malformations are commonest cause of obstructive uropathy in children. Early diagnosis of these defects by prenatal or neonatal ultrasonography is important to prevent secondary effects, e.g.
obstructive uropathy, vesicoureteric reflux, recurrent UTI and occasionally, post-renal failure.Embryology: Urinary tract develops from:
• Mesonephric duct, which arise from lower allantois, just above the terminal end of primitive hindgut, i.e. cloaca; and
• Primitive urogenital sinus, which is formed after division of cloaca by a transverse urogenital septum into anterior urogenital sinus and posterior cloacal sinus (ano-rectal canal).
Ultimately, (a) ureters develop from a ureteric bud of mesonephric duct, (b) trigone of bladder from lower part of mesonephric duct, incorporated into urogenital sinus, (c) urinary bladder (except trigone) from upper part of primitive urogenital sinus, and (d) urethra from lower part of urogenital sinus.
Upper part of mesonephric duct, above the origin of ureteric bud, becomes atretic to form urachus—a ligament between bladder and umbilicus. Urinary tract anomalies may be classified according to the site of
TABLE 21.22: Common urinary tract malformations
• Pelviureteric junction (PUJ) anomalies
- Infundibular stenosis (hydrocalycosis)
- Kinks, bands and strictures
• Ureteric anomalies
- Abnormal number: Agenesis, duplication
- Abnormal location: Ectopic ureters
- Abnormal size: Megaureter, ureterocele
• Urinary bladder anomalies
- Agenesis/hypoplasia
- Exstrophy of bladder (Ectopia vesicae)
• Uretheral anomalies
- Posterior urethral valves
- Hypospadias/Epispadias
obstruction (Table 21.22), of which some common ones are discussed here.
Pelvi-ureteric junction (PUJ) anomalies: PUJ malformations, though more common in males, are commonest cause of obstructive uropathy in females and second commonest in males (after posterior urethral valves).
PUJ anomalies are more common on left side and include a spectrum of defects—intramural defects, e.g. stenosis, persistence of fetal folds, aperistaltic segment, etc. or extrinsic defects, e.g. aberrant vessels, ectopic kidneys.
Clinically, these cases may be picked up on antenatal USG (fetal hydronephrosis) or manifest later with palpable renal mass, hematuria after trivial trauma, recurrent UTI and occasionally, intermittent Dietl's crisis (sudden abdominal pain with renal lump due to ? torsion of pelvis).
Diagnosis rests on USG to show pelvic dilatation without ureteric dilatation, though a DTPA/M AG-3 scan is necessary to exclude milder abnormalities. IVP, though not necessary, shows pelvis dilatation with abrupt cutoff at PUJ.
Treatment: Early surgical repair is indicated in cases with bilateral disease, solitary kidney or severe hydronephrosis with worsening functions. In others, follow-up is advisable as mild to moderate fetal hydronephrosis is known to improve with time.
Posterior urethral valves (PU valves) are the commonest cause of obstructive uropathy in male infants, seen in 1:5000 to 1:8000 children.
Pathogenesis: PU valves are located just distal to the verumontanum at the junction of anterior and posterior urethra. Embryologically, these structures are considered as—(a) extreme development of normal urethral folds and ridges, or (b) remnant of the urogenital membrane. These valves act as a rigid membrane causing obstruction to the urinary outflow, followed by elongation and fusiform dilatation of the proximal urethra. Depending on the severity of lesion, back-pressure leads to variable degree of detrusor muscle hypertrophy, secondary VUR and hydronephrosis.
PUV are also classified as-Type I, which radiate distally from verumontanum and merge to form anterior commissure; Type II, which spread proximally and usually remain undetected; and rare Type III, consisting of a diaphragm with central hole and require retrograde catheterization.
Clinically age of presentation depends on the severity of the obstruction.
Severe cases present in neonatal period with-(a) acute urinary retention with distended bladder, (b) enlarged/ palpable kidneys, (c) urinary ascites and (d) respiratory distress due to associated pulmonary hypoplasia.
Moderate lesions present later with—(a) weak urinary stream, (b) intermittent urinary retention, (c) obstructive uropathy and renal failure, (d) repeated UTI.
Diagnosis: Micturating cystourethrography (MCU) is the investigation of choice for postnatal diagnosis, which shows a dilated posterior urethra, poor flow to distal urethra, residual urine with secondary bladder abnormalities and vesicoureteric reflux.
PU valves may be diagnosed on prenatal USG, which also reveals distended bladder, bilateral hydroureter and hydronephrosis. Oligohydramnios and consequent pulmonary hypoplasia is common.
Antenatally suspected PUV should be confirmed postnatally by USG on Day 3 of life, based on bilateral hydronephroureterosis and thickened distended bladder with dilated proximal urethra (Key-hole sign).
Treatment: Endoscopic fulguration of valve leaflets using a diathermy current, is the treatment of choice in PU valves. However in cases with severe renal insufficiency, initial stabilization with fluid and electrolyte correction, control of urinary infection and temporary drainage of urine (catheter or vesicostomy) is necessary before surgery.
Post-surgery, serial USG monitoring and antimicrobial prophylaxis may be needed in cases with significant vesicoureteral reflux, along with DMSA scan. Bladder dysfunction is common and many cases need intermittent catheterization and bladder augmentation procedures.
Prognosis: While renal functions improve rapidly after surgery, ultimate outcome depends on severity of pre-surgical renal impairment and reduction of serum creatinine levels after surgery. Prognosis is good if postsurgery serum creatinine falls lt;0.8 to 1 mg/dl.
Cases with severe obstructive uropathy or delayed surgery may progress to chronic renal failure, requiring renal transplantation. Urinary incontinence is common even after successful surgery.Prune-Belly syndrome is a characteristic triad of—(a) deficient abdominal muscles, (b) undescended testes
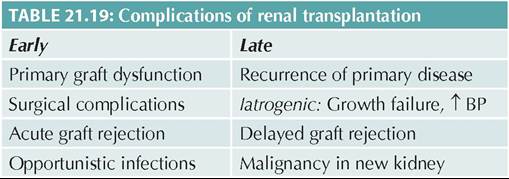
Fig. 21.8: Prune-belly syndrome.
and (c) urinary tract anomalies, e.g. megaureters, megabladder, patent urachus, etc.
Oligohydramnios and pulmonary hypoplasia is common and ~ 95% cases are males. Prognosis depends on the severity of pulmonary hypoplasia and renal dysplasia. While ~1/ 3rd cases die in utero or within few weeks, another 1 / 3rd progress to end-stage renal disease (Fig. 21.8).
Bladder exstrophy is a rare (1:40,000 births) malformation with variable severity, ranging from merely epispadias or small vesicocutaneous fistula in abdominal wall, to complete ectopia vesicae.
Embryologically, these anomalies indicate complete/ partial failure of urogenital septum to connect with cephalad part of cloacal membrane.
In classic cases, bladder protrudes from abdominal wall with exposed mucosa and complete epispadias. Pubic rami and recti muscles are widely separated and anus is anteriorly displaced. Undescended testes and inguinal hernia are common.
Management involves multi-stage reconstructive surgery including closure of exstrophic bladder at birth, correction of epispadias after infancy and lastly, the reconstructive surgery to achieve continence.
However, post-surgical complications are common and include—(a) urinary incontinence, (b) recurrent UTI, (c) sexual dysfunction, and (d) risk of bladder cancers in later life.
21
Hypospadias is the commonest congenital anomaly of urethra, characterized by abnormal urethral meatus opening on the ventral surface of penile shaft rather than on the glans-tip, with or without chordee (abnormal ventral curvature of the penis) and underdeveloped prepuce.
Although usually an isolated defect, many cases are associated with other anomalies of genitourinary tract, e.g. undescended testis, inguinal hernia, vesicoureteric reflux, etc.
TABLE 21.23: Classification of hypospadias
Anterior hypospadias (80-85%)
• Glandular
• Coronal
• Sub-coronal or distal penile
Middle hypospadias (10-15%)
• Mid-penile
Posterior hypospadias (3-6%)
• Proximal penile
• Peno-scrotal
• Scrotal
• Perineal
Fig. 21.9: Hypospadias.
Incidence is estimated to be ~1:250-500 male births, more common in newborns with in utero estrogen exposure. Clinical presentation of hypospadias depends on the location of the urethral orifice (Table 21.23). Depending on the severity of abnormality, these cases present with-(a) asymptomatic cosmetic defect, (b) abnormal urinary stream with ventral deflection or splaying, (c) sexual dysfunction due to chordee, and (d) features of co-anomalies, e.g. obstructive uropathy (Fig. 21.9).
Diagnosis is clinical, though radiological evaluation of whole urinary tract is necessary to exclude coexisting urinary tract and renal anomalies. Cases with severe hypospadias should also be differentiated with ambiguous genitalia by karyotyping.
Management aims to correct cosmetic as well as function deformity with surgical repair, best started at 6-12 months of age. Although the type of surgery depends on the location of meatus, general principles include—
(a) meatoplasty, (b) urethroplasty, and (c) correction of chordee.
Complication rate of surgery varies from 5 to 15%, including urethrocutaneous fistula, meatal stenosis, urethral strictures/diverticula and wound infection.
Epispadias, i.e. urethral meatus opening on the dorsal surface of penis, is relatively less common than hypospadias and frequently associated with short, upturned penis and urinary incontinence.
Based on the site of the meatal opening, epispadias is classified as—glandular, penile or penopubic.
In females, similar displacement of urethral meatus may present with bifid clitoris, flattening of the mons and labial separation, classified as—minimal (Patulous urethral opening), intermediate (dorsally split urethra along most of its length) or severe (complete urethral cleft with incompetent sphincter).
Management depends on the severity, with surgical repair involving meatostomy, urethrostomy and release of dorsal chordee (Cantwell-Ransley repair or corpora-corporal anastamosis), usually at the age of 2 years.
21.14