CELL DEATH PATHWAY DURING ACUTE HIV-1 INFECTION
Acute HIV-1 infection causes massive death in virus-producing cells. This can be readily observed in highly infected cultures of transformed cell lines or nontransformed peripheral blood CD4+ T cells.11,12 What is the basic mechanism of direct viral killing? Multiple pathways could lead to pathological cell death, including forms of programmed cell death, typically apoptosis, and traumatic death by injury called necrosis.
Signals might be intertwined enough to ultimately lead to multiple death pathways downstream of the signaling cascade, which could result in the hallmarks of both pathways in infected cells. Therefore, data have to be carefully examined in order to draw firm conclusions.Modes of Cell Death: Apoptosis vs. Nonapoptotic Cell Death
Apoptosis refers to a programmed cell-death pathway that exhibits distinct morphological changes catalyzed by a family of cysteinyl aspartate-specific proteases called caspases. The activation of caspases leads to characteristic biochemical changes, including chromatin condensation, DNA fragmentation, externalization of phosphatidylserine on the plasma membrane, and the exposure of a specific mitochondrial membrane protein. These changes are detectable by such biochemical techniques as Terminal dUTP Nuclear End Labeling (TUNEL) assays, Annexin V staining, and APO2.7-antibody staining, (detailed discussion can be found in Chapter 3).23-25 In addition to the biochemical changes, caspase-activated death has a characteristic morphological appearance on electron microscopy. The cell nucleus becomes smaller and electron dense, the cytoplasm condenses, and the cell membrane is intact but may have a ruffled appearance—all of which are easily distinguished from other modes of cell death and preventable by caspase inhibitors.
Another major form of cell death involves necrosis.
In contrast to apoptosis, necrosis exhibits loss of plasma membrane integrity and cellular swelling followed by lysis due to traumatic damage. Unlike apoptosis, the contents of the cell are released upon lysis, which could be advantageous to stimulation of immune responses against pathogens. Although condensation of chromatin does not occur in necrosis, DNA degradation is observed at a later stage, resulting in DNA smearing on gel electrophoresis, similar to a DNA laddering effect during apoptosis.26,27 These two major forms of death are readily distinguished morphologically by electron microscopy. Unlike apoptosis, necrotic cells become enlarged with fragmentation of the plasma membrane, decreased density of the cytoplasmic region, and dissociation of the nucleus and other organelles. Biochemical methods are also frequently used to distinguish apoptotic vs. necrotic changes in dying cells. Perhaps the easiest test is the detection of the fluorescent dye propidium iodide (PI) binding to the genomic DNA, which occurs only when the plasma membrane integrity is lost. Early PI staining will occur in necrosis but not apoptosis. However, many of the commonly used biochemical assays do not clearly distinguish apoptosis from necrosis.Efforts to understand the mechanism of CD4+ T cell depletion led to the extensive investigation of the molecular mechanism of cell death during HIV-1 infection, unfortunately resulting in conflicting hypotheses. Large numbers of studies proposed apoptosis to be the major mode of viral- induced cell death based on the appearance of apoptotic features in infected cultures.28,29 However, significant data challenged the view that apoptosis is the mechanism of viral-mediated CD4+ T cell destruction. For example, many experiments designed to disrupt the apoptosis pathway failed to abrogate viral-induced cell death. A number of other studies could not establish a requirement for the apoptosis-inducing caspases in HIV-1 killing.6,7,11,12,30,31 The surface expression of a key apoptosis receptor in T cells, Fas/APO-1/CD95, and its ligand, FasL, was not upregulated by HIV-1 infection in PBMCs.10 Studies also demonstrated that viral-induced death could bypass the requirement of the Fas death pathway.10,11 Over expression of antiapoptotic proteins, Bcl-2, Bcl-xL, and adenovirus E1B 19K did not significantly alleviate cell death in infected populations,11,32,33 casting further doubt on apoptosis as the major death pathway for HIV-1 killing.
Moreover, other studies were unable to establish a correlation between the level of apoptosis and disease progression.16These discrepancies also raise questions regarding the validity of in vitro systems used to detect apoptosis in some of the experiments and the conclusions made using them. It is well documented that biochemical changes observed in apoptotic cells are not always specific and, therefore, biochemical assays used to investigate apoptosis do not always detect apoptosis exclusively (discussed in Chapter 3). A typical example is the TUNEL assay used to detect nonspecific DNA fragmentation. DNA fragmentation is a feature of cells undergoing apoptosis, but the assay may also detect DNA degradation observed during necrosis in T cells.9,26,27,34-36 Another widely used assay for apoptosis, Annexin V staining, detects damaged cells by exposing phosphatidylserine, irrespective of the mode of death.37,38 Due to these complications, individual biochemical assays do not necessarily make a clear distinction between apoptosis and necrosis when used alone. Additionally, the interpretation of many studies in the literature became more difficult when cell death was examined in assay systems that did not distinguish infected from uninfected cells. Apoptotic features as well as cell death observed in those studies do not always correlate in kinetics or magnitude with the degree of viral infection, which makes it difficult to make inferences on virus-induced cell death.9,39,40 These complications raise the question of whether cell death is due to a specific and direct effect of HIV-1 infection rather than general adverse conditions in cell culture associated with infection.
The analysis by multiple distinct biochemical tests for apoptosis provided persuasive evidence that necrosis rather than apoptosis was the primary mode of death. Importantly, electron micrographs and video imaging of infected cells confirmed the absence of prominent apoptotic features, such as nucleus condensation and fragmentation, in infected PBMCs and T cells from HIV-positive patients (Figure 17.1).9,12,41 The involvement of apoptosis in provirus-expressing T cells was also investigated by treating cells with an apoptosis inhibitor, zVAD-fmk.8,10,11 Whereas general Fas-induced apoptosis was blocked in drug-treated cells, virally induced death in infected cells was not impaired by this treatment, indicating that apoptosis is dispensable for direct HIV-1 killing of T cells.
This result was confirmed by experiments using T cell lines deficient in primary apoptosis factors, such as caspase-8, FADD, and RIP.42,43 Experiments in CD4+ T cells from patients with autoimmune lymphoproliferative syndrome (ALPS).10 ALPS is a disorder caused by a genetic defect in the apoptosis signaling pathway induced by the death receptor Fas that leads to a specific resistance to apoptotic stimuli.44 Patients harboring mutations in the Fas gene have significantly reduced susceptibility to Fas-induced apoptosis. As expected, direct viral killing was not prevented in cells lacking the primary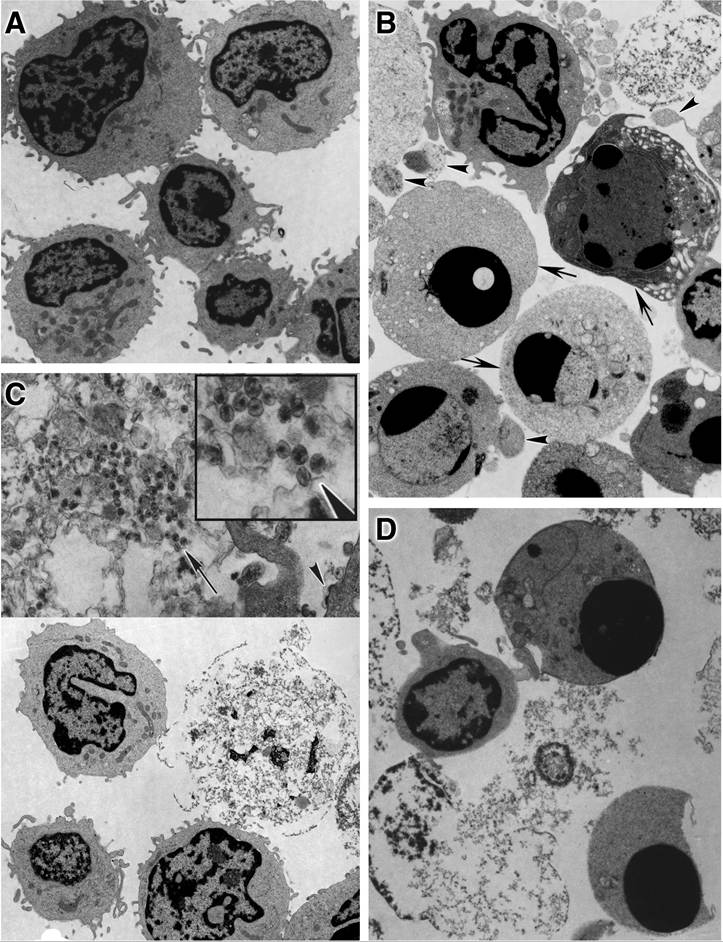
FIGURE 17.1 Electron micrographs of activated CD4+ T lymphocytes comparing cell death by HIV-1 infection with staurosporine-induced apoptosis. Activated CD4+ T lymphocytes were either infected with NL4-3 Env-virus or mock infected for 9 days and then were treated with staurosporine (1 i.ig/niL) for 4 h. (A) Mock- infected cells. (Original magnification ? 4900.) (B) Mock-infected cells with staurosporine added. (Original magnification ? 5400.) (C) HIV-1-infected cells. (Lower half, original magnification ? 4900; upper half, original magnification ? 17,000.) (D) HIV-1-infeceted cells with staurosporine added. (Original magnification ? 7200.) In (B), arrows indicate cells with compacted chromatin, a hallmark of apoptosis; arrowheads indicate budding apoptotic bodies. In (C), the arrowhead indicates budding virions; the inset in (C) is a 2.2-fold magnification of the region indicated by the arrow and reveals mature retroviral particles within the debris of a necrotic cell. (From Lenardo, M.J., J. Virol., 76, 5082, 2002. With permission.)
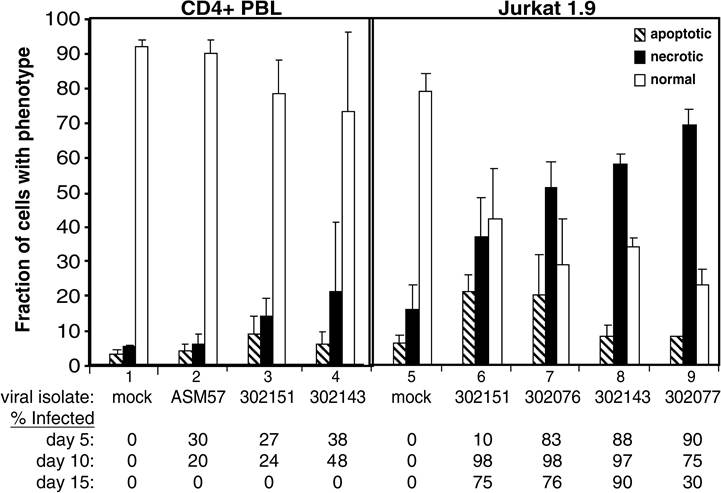
FIGURE 17.2 Quantification of apoptosis and necrosis in peripheral blood CD4+ T lymphocytes and Jurkat 1.9 cells infected with HIV-1 primary isolates.
Cells were either infected with primary isolates of HIV-1 or mock infected; after 10 days, cells from each sample were analyzed for morphological features of apoptosis or necrosis from transmission electron micrographs of stained microscope slides. Samples 1 to 4 represent peripheral blood CD4+ T cells; samples 5 to 9 represent Jurkat cells. The primary isolates used in each infection were as follows: samples 1 and 5, mock-infected controls; sample 2, ASM 57 virus from a long-term nonprogressor; samples 3 and 6, dualtropic virus 302151; samples 4 and 8, dualtropic virus 302143; sample 7, dualtropic virus 302076; and sample 9, dualtropic virus 302077. The bottom of the figure provides the fraction of infected cells, as determined by staining of p24 and flow cytometric analysis for the indicated time points. (From Lenardo, M.J., J. Virol., 76, 5082, 2002. With permission.)apoptosis factors or in those from ALPS patients.10,11,42,43 Infection with HIV-1 apparently causes direct cell death in infected CD4+ T cells, which is not dependent on the apoptotic pathway.
It should be noted that a small fraction of cells in tissue culture infections is often found to exhibit apoptotic markers in vitro.9~11 One possible explanation is that infected cells show increased vulnerability to apoptosis, possibly due to compromised metabolic conditions within infected cells. Many reports have documented that infected PBMCs and T cell lines become more sensitive to apoptotic stimuli,29,45-47 which could contribute to the presence of apoptotic features among infected cells. The mechanism by which this increased susceptibility occurs is not well understood. Nonetheless, the fraction of cells expressing apoptosis markers is minimal, as discussed above, and does not account for massive cell death induced by HIV-1 infection.
To address the issue of direct necrosis vs. bystander death, recent studies have employed a single-cell killing system that allows a clear distinction between infected and uninfected cells.
Laboratory strains of HIV-1 were engineered to incorporate a tag, such as placental alkaline phophatase (PLAP), green fluorescent proteins (GFPs), or a mouse heat-stable-antigen∕CD24 (HSA) marker, to monitor infection.10-12,19 These systems also allowed the quantitative cell-by-cell measurement of proviral expression by flow cytometry. Careful analyses of provirus/reporter-expressing cells demonstrated that massive cell death occurred in infected cells rather than in bystander cells and that infected cells did not exhibit apoptotic hallmarks such as DNA fragmentation and extracellular flux of phosphatidylserine (Figure 17.1).11,12 Taken together, evidence strongly indicates that HIV-1 cytopathicity does not involve apoptosis; rather, it predominantly occurs via necrosis. However, this conclusion is based on careful studies carried out in vitro and must be further validated by studies of HIV infection in vivo.Molecular Mechanism of Nonapoptotic/Necrotic Cell Death
Mitochondrial Dysfunction
A variety of agents that cause lethal cellular damage could result in necrosis or apoptosis via mitochondrial dysfunction. These agents include pathogens and oxidative stress from nitric oxide radicals and reactive oxygen species (ROS). They initiate a death signal by causing drastic changes in the permeability of the mitochondrial inner membrane, which could subsequently trigger the release of proapoptotic molecules such as cytochrome C and Smac/Diablo. This process, called mitochondrial permeability transition (MPT), can lead to either apoptosis or necrosis.48-51 Studies suggest that the switch between necrosis and apoptosis during mitochondrial dysfunction is modulated by intracellular ATP concentration and caspase activation.49,52 During MPT, the permeability of the mitochondrial membrane to ions and solutes increases due to the unregulated opening of MPT pores. Subsequent to the MPT pore opening, the mitochondrial transmembrane potential (∆ψm) is perturbed. This is accompanied by the generation of superoxides, uncoupling of oxidative phosphorylation, and ATP depletion, which results in MPT-dependent necrosis.49-53
HIV-1 is reported to initiate the loss of ∆ψm upon infection. Three HIV-1 proteins, gp120, Tat, and Vpr, are documented to act as causative agents.9,14,54-57 The association of soluble gp120 and CXCR4 has been reported to cause opening of the MPT pore followed by the rapid reduction in ∆ψm and, eventually, cell death due to mitochondrial dysfunction.56,58 In addition to the direct effect, Env-mediated syncytia is also reported to trigger mitochondrial membrane permeabilization and disruption in HeLa cells expressing CD4 and CXCR4.59 However, it is still controversial whether gp120-induced cell death occurs by apoptosis or necrosis.21,56,58 Tat is another viral protein proposed to induce MPT and cell death. The constitutive expression of Tat by stable transfection in a T leukemia cell line has been demonstrated to cause a loss of ∆ψm and downregulate mitochondrial superoxide dismutase (SOD2), resulting in the accumulation of ROS and mitochondrial dysfunction.60,61 Intriguingly, Tat translocates from the nucleus and cytoplasm to mitochondria concomitantly with the ∆ψm disruption,62 which supports a role of Tat in MPT. During Tat-induced mitochondrial dysfunction, cell death was not abrogated by an inhibitor z-DEVD-cmk for caspase- 3, the most important effector caspase.62 This result suggests that Tat-mediated mitochondrial dysfunction leads to necrotic death. In addition to Env and Tat, Vpr is also suggested to cause death through the mitochondrial pathway. A fraction of Vpr is reported to physically associate with adenine nucleotide translocase (ANT) in mitochondria. This interaction is thought to form large composite ion channels, which facilitates dissipation of ∆ψm, leading to cell death.63,64 A recent study reported an interaction between Vpr and ANT, and the loss of ∆ψm is attributed to the C- terminal region of Vpr (a.a. 52-96), especially R73 and R80 residues,64 which are also important for the cell-cycle arrest property of Vpr.65,66 Intriguingly, HIV-1 carrying a mutation at Vpr∕R88 (R88A) is reported to be significantly less cytopathic in a Jurkat T cell line,65,67 suggesting a correlation between Vpr-mediated cytopathicity and mitochondrial dysfunction. Similar to Tat, Vpr- induced MPT and cell death are not inhibited by a caspase inhibitor, zVAD-fmk, or the knockout of mitochondrial apoptosis-inducing factor Apaf-1.68 These data suggest that Vpr mediates nonap- optotic cell death through the mitochondrial pathway. HIV-induced mitochondrial dysfunction is consistent with the observations that T cells from patients undergo changes in mitochondrial morphology, a reduction of ∆ψm, and enhanced ROS generation.54,55,57 In contrast, long-term nonprogressors have a low incidence of reduced ∆ψm,69 suggesting a correlation between mitochondrial dysfunction and HIV-1 pathogenesis. Further studies are necessary to elucidate the detailed mechanism by which HIV-1 causes the loss of ∆ψm.
Cell-Cycle Arrest
HIV-1 infection causes cell-cycle arrest at G2/M phase and Vpr functions as a primary mediator of cell-cycle blockades (Vpr is discussed in Chapter 7). Several groups investigated the correlation between cell-cycle arrest by Vpr and viral-induced cell death. However, the studies failed to reach a consensus (details discussed below).65∙67∙70-74 In addition, the mode of death is not yet clearly established in cells arrested by Vpr. Further investigation is necessary to determine whether cellcycle arrest is a causative factor of cell death.