CELL DEATH BY SUICIDE
The importance of mitochondria in initiating apoptosis was realized in 1996, when Liu and colleagues demonstrated that cytochrome c, a component of the electron transport chain, could initiate apoptosis when released into the cytosol.41 Upon release from mitochondria, cytochrome c forms a complex with Apaf-1 and ATP to activate caspase-9, initiating the intrinsic caspase cascade.
Subsequent studies showed that mitochondrial dysfunction and loss of membrane integrity cause release of multiple proteins, which contribute to the cell death program. For example, Smac/DIABLO and Omi/HtrA2 inhibit IAP family proteins to reinforce caspase-9 activation. EndoG and AIF are proposed to be components of caspase-independent pathways leading to nuclear fragmentation.The mitochondrial pathway of apoptosis is initiated by internal cellular stress, such as DNA damage, cytokine deprivation, hypoxia, and other stimuli. Controversy exists over the mechanism by which mitochondria lose integrity and release their contents into the cytosol. Some hypotheses suggest that mitochondrial dysfunction leads to matrix swelling and eventual rupture of the outer membrane. Others propose the formation of specific pores large enough to release intermembrane components. Under either model, the importance of Bcl-2 family members in regulating mitochondrial membrane integrity is paramount. Another input for regulation occurs at the transcriptional level, by p53, and at the posttranslational level, by survival kinase signaling pathways.
Bcl-2 Family
The first molecularly characterized example of an apoptotic genetic defect in cancer was the t(14:18) chromosomal translocation found in follicular lymphoma patients that juxtaposes the Bcl-2 gene with the immunoglobulin enhancer.42 Later work revealed that the Bcl-2 gene product promotes oncogenesis by a novel mechanism: rather than inducing cell proliferation, Bcl-2 inhibits the normal programmed cell death of B cells.43,44 The mechanism by which antiapoptotic proteins Bcl-2 and Bcl-xL promote survival is not completely clear, but one hypothesis involves suppression of the activation of proapoptotic members Bax and Bak.45 Bax and Bak can initiate apoptosis when they oligomerize at the mitochondria,46 disrupting membrane integrity to release cytochrome c, or at the endoplasmic reticulum,47-48 causing disruption of Ca2+ homeostasis.
Alternatively, Bcl-2 and Bcl-xL were proposed to preserve mitochondrial membrane integrity by promoting the exchange of metabolic substrates across the mitochondrial membrane and allowing maintenance of respiration.49Whereas these main apoptotic effectors contain multiple Bcl-2 homology (BH) domains, other family members share only the BH3 region. Subsequent investigations defined distinct roles for individual proapoptotic BH3-only proteins,50 with functions that can be divided into two groups based on binding specificity. Bid-like domains exert proapoptotic functions by activating Bax and Bak, and Bad-like domains sensitize cells to apoptosis by occupying the binding pocket of antiap- optotic members Bcl-2 or Bcl-xL.51 Therefore, the ratio of proapoptotic to antiapoptotic Bcl-2 family proteins determines the status of mitochondrial permeability and cell survival (Figure 27.1).
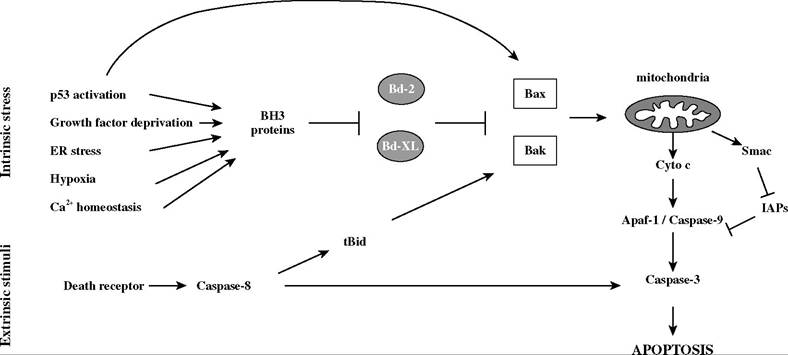
FIGURE 27.1 Regulation of apoptosis by the Bcl-2 family proteins. After cell autonomous stress, the balance between BH3-only and Bcl-2 family proteins regulates mitochondrial membrane permeability and apoptosis. In response to external death receptor signaling, cleavage of Bid amplifies the mitochondrial death pathway.
Induction of Cell Death with Bcl-2 Antisense or BH3-Mimetic Strategies
Bcl-2 overexpression can cause resistance of cells to various apoptotic stimuli, including chemotherapy, hypoxia, radiation, Ca2+ overload, ceramide, and growth factor deprivation. One approach to modulating the ratio of antiapoptotic to proapoptotic proteins is to target the expression of antiapoptotic molecules with antisense strategies. For example, antisense oligonucleotides that reduce Bcl-2 expression (such as Genasense) effectively sensitize cancer cells to chemotherapy and are being used in clinical trials for hematologic malignancies52,53 and several solid tumors.54,55
As a promising alternative strategy to antisense approaches, structural studies of BH3 dimerization domains56 led to the discovery of small molecule inhibitors that prevent protein-protein interactions of Bcl-2 family members.
Initial studies used cell-permeable peptide inhibitors to bind Bcl-2 and sensitize cancer cells to apoptosis.57 The first small molecule inhibitors, BH3I-1 and BH3I-2, were discovered by screening a chemical library for compounds that disrupted the interaction between Bcl-xL and the Bak BH3 peptide.58 Another study serendipitously found that Antimycin A competes with Bak for interaction with Bcl-xL or Bcl-2.59 Several other studies utilized synthetic BH3-mimetic peptides60 or naturally derived chemicals61 to induce mitochondrial membrane permeabilization by blocking Bcl-2 or Bcl-xL function.Prevention of Cell Death by Maintenance of Mitochondrial Membrane Integrity
In contrast to cancer, diseases characterized by excessive cell death would be improved by therapies that enhance the activity of survival proteins such as Bcl-2 and Bcl-xL. Bcl-2 overexpression was shown to prevent cell death in multiple animal models of neurodegeneration, ischemic injury, and septic shock.62-64 Besides gene therapy, an alternative strategy to increase protein expression can be achieved by using the HIV Tat protein conjugation system for intracellular delivery of pep- tides.65,66 Although the mechanism of Tat transduction across the membrane is not entirely clear, it is thought to involve lipid-raft-dependent macropinocytosis.67 Tat-mediated protein transduction of Bcl-xL showed protective effects against cell death in a rodent model of global ischemia.68
As an alternative to altering expression of antiapoptotic proteins, mitochondrial membrane integrity can also be preserved with drugs that block their activity either directly or through upstream signaling pathways. For example, immunosuppressants such as FK506 and cyclosporine A are emerging as clinically useful therapeutic agents for neurodegenerative diseases. Because their immunophilin receptors are expressed 10- to 100-fold higher in central and peripheral nervous system tissue than in immune tissue, they can be protective in neurons at low doses without affecting the immune system.
The neuroprotective effect of FK506 was first reported in ischemic brain injury69 and later found to function by blocking calcineurin. Although both FK506 and cyclosporine A can block calcineurin,70 cyclosporine A can also inhibit the mitochondrial matrix prolyl-isomerase cyclophilin D and prevent activation of the mitochondrial permeability transition pore. Because FK506 lacks the ability to bind cyclophilin D, cyclosporine A provides more neuroprotective effects in animal models of traumatic brain injury,71 ischemia,72 and neurodegeneration.73 Other drugs might preserve mitochondrial membrane integrity by directly disrupting Bax or Bak function. For example, dibucaine and propranolol were proposed to block Bax-induced changes in lipid structure.74 Further investigation into the mechanism of outer-membrane permeabilization induced by BH3 death domain protein-Bax interaction will likely lead to the development of more specific antiapoptotic therapeutics relevant for neuroprotection.p53 Status
The tumor suppressor p53 is another major point of control in the initiation of apoptosis, and loss of p53 function in tumor cells contributes to resistance to chemotherapy and radiation. p53 stability and subcellular localization are regulated by multiple posttranslational modifications and proteinprotein interactions in response to stresses including DNA damage, activation of oncogenic signaling, and hypoxia.75 p53 stabilization leads to cell cycle arrest or apoptosis and exerts these effects in both transcription-dependent and transcription-independent ways. p53 directly activates multiple pathways through transcription of apoptotic genes, including some death receptors, cytosolic proteins Bid and Apaf-1, and mitochondrial effectors Bax and Bak. p53 further promotes apoptosis through transcriptional repression of antiapoptotic genes, such as Bcl-2 and survivin.76 Early studies also suggested a transcription-independent role for p53.77 Recent reports found that p53 localizes directly at the mitochondria, though further research is required to understand the significance of this finding.78-81
Induction of Cell Death by Restoration of p53 Function
Restoration of disrupted apoptotic pathways can be accomplished by multiple methods.
If a gene product is lost or mutated, as for example p53, gene therapy may provide direct restoration of expression of the missing wild-type protein. The most common approach taken for delivery has been use of an adenoviral vector.82 Intratumoral injections have shown some success in inducing tumor regression. Several approaches are currently in clinical trials, including a construct from Introgen Therapeutics (INGN201) for head and neck cancer83 and a construct from Schering-Plough (SCH58500) for advanced ovarian cancer.84,85 Another construct, called Gendicine, was recently approved for commercial use in China and shows significant tumor regression in combination with radiotherapy for head and neck squamous cell carcinoma.86Another genetic approach to target cancer cells lacking functional p53 is the use of oncolytic viruses. This strategy takes advantage of the fact that adenoviruses must inactivate p53 in order to replicate in cells. Wild-type adenovirus accomplishes this inactivation through the viral protein E1Bp55, which binds and inactivates p53. A virus lacking E1Bp55, therefore, selectively replicates in and causes lysis of cancer cells that lack p53.87 This approach is being used clinically with the drug ONYX-015, which showed success in combination with chemotherapy in phase II trials when injected into squamous cell carcinomas of the head and neck88 and in phase I studies of oral dysplasia.89 Although these approaches seem promising for topical application or intratumoral delivery of solid tumors, systemic gene therapy poses additional challenges, because feasibility is limited by efficiency of transgene expression. Furthermore, the overall safety of adenoviral vectors remains an issue.
In addition to gene deletion, a common mechanism by which p53 function is lost is through development of loss-of-function point mutations. To target mutant p53 in cancer, one strategy was to use small molecules that restore p53 function.
CP-31398 is a drug that was found through a chemical screen of compounds that stabilize the active conformation of the DNA binding domain of p53, rescuing the conformation and function of mutant p53 in tumor cells.90 Subsequent studies on the mechanism by which this drug causes apoptosis yielded conflicting results, with some finding restoration of p53 function to be responsible for cell death and others citing nonspecific toxicity.91,92 Prima1 is another small molecule compound that restores the DNA binding ability of mutant p53,93 and follow-up studies on its specificity are forthcoming. Recent reports suggested that small molecule drugs might also activate transcription-independent functions of p53 in inducing apoptosis.78As an alternative to gene therapy and small molecule drugs, a more controversial approach to restoring p53 function involves systemic administration of a transducible p53-activating peptide. A peptide derived from the C-terminal end of p53 was recognized as an activator of endogenous wild-type p53 and some mutants.94 Because this peptide functions by restoring DNA-binding ability, it can induce apoptosis in tumor cells containing wild-type or DNA-binding mutants of p53 but not in cells deficient for p53 or containing structural mutants. A recent study showed that delivery of this peptide into cells by Tat-mediated transduction can selectively activate p53 and induce apoptosis in cancer cells but not normal cells.95 Importantly, this approach showed in vivo efficacy in animal models of human terminal malignancy, such as peritoneal carcinomatosis and peritoneal lymphoma. Whereas current clinical use of macromolecular agents is limited to extracellular factors, the evidence from this study suggests that delivery of intracellular targets by Tat-mediated transduction might be another viable therapeutic option.
Induction of Cell Death by Activating Wild-Type p53
Recent attempts at targeting wild-type p53 to induce cell death showed that small molecule drugs can be designed to specifically disrupt the interaction between p53 and its negative regulator MDM2. MDM2 functions as an E3 ubiquitin ligase to promote p53 nuclear export and degradation and specifically binds to a 15-amino-acid segment of the p53 transactivation domain to block transcriptional activity.96 In turn, the tumor suppressor ARF negatively regulates MDM2 by sequestering it in the nucleolus. Abnormalities of these upstream regulators tend to occur exclusively of each other97 and of p53 mutations, demonstrating that the p53/MDM2/ARF pathway can be disrupted at multiple points to functionally inactivate p53 activity. Blockade of the MDM2-p53 interaction might be a particularly useful therapy in tumors where MDM2 is overexpressed, such as in some non-small cell lung carcinomas, breast cancers, and brain tumors98,99 or where ARF is deleted or mutated, such as in melanomas and colon cancer.100
Studies investigating inhibition of MDM2 established that disrupting its interaction with p53 can lead to a p53 response and tumor growth inhibition. However, inhibition of MDM2 expression with antisense oligonucleotides suppressed growth of both wild-type and mutant-p53-containing cancer cells in animal studies101,102 and in clinical trials.103 The possibility that small molecules might effectively target this protein interaction arose when structural studies revealed that only three amino acid side chains from p53 were responsible for its interaction with MDM2.96 In order to identify compounds that could specifically disrupt this interaction and elicit p53-dependent apoptosis, Vassilev et al.104 screened a library of synthetic chemicals for the ability to displace recombinant p53 from MDM2. They identified a series of compounds, termed nutlins, that specifically block this interaction as verified by crystal structure analysis. These compounds induced cell cycle arrest and apoptosis in human cancer cell lines with wild-type p53 but not mutant p53 and effectively suppressed growth of human tumor xenografts in nude mice.
Wild-type p53 stabilization might also be achieved by blocking its interaction with the negative regulator, Parc.105 Parc was recently identified as a cytoplasmic anchor for p53, which controls p53 subcellular localization and function. Many neuroblastoma cell lines that exhibit a primarily cytosolic localization of wild-type p53 were found to overexpress Parc. Knockdown of endogenous Parc expression in these cells by RNA interference caused apoptosis by inducing redistribution of p53 to the nucleus and sensitized neuroblastoma cells to genotoxic stress. The feasibility of targeting this interaction with small molecule inhibitors depends on specificity and awaits further structural studies.
Survival Kinases
Another mechanism by which cancer cells evade apoptosis is through constitutive activation of cell survival signaling cascades, including the PI3K/Akt, MAPK, and NF-κB pathways (Figure 27.2).
PI3K/Akt/mTOR
The phosphatidylinositol-3-kinase (PI3K) pathway plays a major role in signaling for growth factors such as interleukin-2, interleukin-3, platelet-derived growth factor (PDGF), and insulin-like growth factor (IGF). Upon growth factor binding, receptor tyrosine kinases (RTKs) dimerize and autophosphorylate tyrosine residues in their cytoplasmic tails. This signal can activate PI3K directly or can recruit adapter proteins that activate the Ras small GTPase to activate PI3K. Once activated, PI3K phosphorylates phosphatidylinositide-4,5-bisphosphate (PIP2) to phosphatidylinositide-3,4,5- triphosphate (PIP3), which acts as a second messenger to activate downstream kinases, such as the
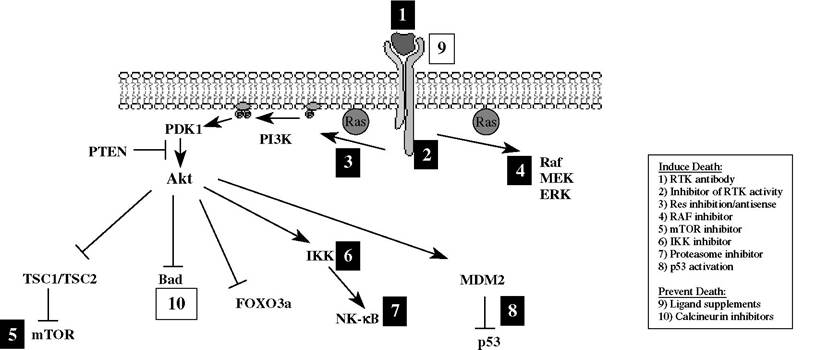
FIGURE 27.2 Therapeutic modulation of survival kinase signaling pathways. Strategies to induce cell death (black boxes 1-8) and to prevent cell death (white boxes 9-10) are highlighted. Survival factors or transforming events activate PI3K signaling, which generates phosphoinositides that recruit Akt to the plasma membrane. After activation, Akt phosphorylates numerous effector molecules that promote cell survival. Current therapeutics target both upstream and downstream components of this pathway to modulate cell survival.
serine/threonine kinase Akt. Akt promotes cell survival by multiple mechanisms, including phosphorylation and inactivation of the proapoptotic BH3-only protein Bad,106-107 inhibition of Forkhead transcription factors that regulate Bim and Fas expression,108 and inhibition of the tuberous sclerosis complex proteins (TSC1/TSC2) that block mTOR activation.109 In addition, Akt promotes glucose uptake and glycolysis upon growth factor stimulation, leading to sustained growth and proliferation. Akt’s role in maintaining substrate availability for mitochondrial respiration was proposed to protect mitochondrial membrane integrity and prevent apoptosis.110,111
Constitutive Akt signaling can occur through amplification of one of the three Akt genes or through loss of its negative regulator, the phosphatase and tensin homologous on chromosome 10 (PTEN). PTEN is a dual specificity protein and lipid phosphatase that degrades PIP3 into PIP2, terminating the signal of PI3K and preventing Akt activation.112 Second only to p53 mutations, loss of PTEN is one of the most frequent aberrations seen in cancer and is found in the majority of glioblastomas and prostate cancers.
PI3K∕Akt∕mTOR Targeted Therapeutics
Deregulation of protein kinase cascades may occur by amplification or overexpression of RTKs (i.e., HER2∕Neu in breast cancers) or by activating point mutations in cytoplasmic signaling kinases (i.e., BRAF in melanoma). RTKs were targeted with monoclonal antibodies that either directly block ligand binding or prevent receptor dimerization. Another approach to kinase inhibition involves small molecule antagonists that directly target the kinase domain, acting as either ATP competitors or substrate mimetics. Alternatively, a more indirect strategy was to target molecular chaperones, such as Hsp90, in order to reduce kinase stability and promote degradation. Antisense oligonucleotides are being investigated for inactivation of H-ras, a critical signaling molecule between RTK and PI3K/Akt activation.113
An early example of a specific RTK inhibitor to become FDA approved was Herceptin, a monoclonal antibody against the HER2 receptor tyrosine kinase for the treatment of breast cancer.114,115 Imatinib mesylate (Gleevec), a small molecule kinase inhibitor that targets Bcr-Abl,116 c-Kit, and PDGFR, was approved for the treatment of chronic myelogenous leukemia in 2001117 and also showed efficacy in gastrointestinal stromal tumors.118 Another drug currently in use is Iressa, a small molecule inhibitor of the epidermal growth factor (EGFR), for treatment of non-small cell lung carcinoma.119
RTK inhibitors likely achieve some of their antitumor effects by targeting upstream signaling of the PI3K pathway. However, their success is hampered in tumors that harbor activating mutations in downstream signaling components, such as Akt or PTEN.120 Direct inhibitors of PI3K, such as LY294002 and wortmannin, were extensively used in research studies but lack specificity among various isoforms of PI3K. Pending development of structure-based specific inhibitors, alternative strategies to directly inhibit PI3K include peptido-mimetics of the p85 SH2 binding site for phosphotyrosine121 or inhibitors that disrupt the Ras-PI3K interaction.122 Alternatively, restoration of PTEN function by gene therapy might provide growth-suppressive effects in appropriate tumors.
Specific targeting of downstream effectors of Akt responsible for cell growth and survival might allow tumor targeting while sparing other functions of Akt not involved in tumorigenesis, such as insulin signaling in metabolism. Of these, the best-studied therapeutic target is mTOR (mammalian target of rapamycin).123 mTOR signaling controls cell growth by regulating translation initiation. In response to nutrients and signal transduction, mTOR phosphorylates 4EBP1, activating capdependent translation, and S6K, inducing ribosome biogenesis. Its direct inhibitor rapamycin was one of the first natural antiproliferative agents to be discovered. Clinical studies are underway for the related compounds RAD001 and CC1779, with the latter in phase III trials for treatment of breast, pancreatic, brain, and renal cancers.
In parallel to the Akt/mTOR signaling pathway, the Pim-2 kinase was shown to promote survival signaling by cytokines.124 Although its importance in human cancer is still under investigation, Pim-2 might represent an important target for tumors that show resistance to rapamycin treatment.
MAPK Signaling and BRAF
Multiple survival kinases were demonstrated to be activated by single-point mutations to render a signaling cascade active. One of the most exciting targets recently identified is BRAF, which was found to be mutated in 67% of melanomas.125 BRAF is an effector of the MAPK cascade, which involves sequential activation of Ras/Raf/MEK/ERK to ultimately alter gene transcription in favor of proliferation and tumor invasion when corresponding growth suppressors (such as p53 and Arf) are absent. The most common activating mutation found in BRAF was a valine-to-glutamate substitution at residue 599. Melanoma cells harboring this point mutation are dependent on its activity for survival, as suppression of mutant BRAF by RNA interference induces apoptosis.126 A small molecule inhibitor against BRAF is currently in clinical trials for treatment of patients with melanoma and other malignancies (Bay 43-9006, in Phase III).127 One possible hurdle for this drug will be specificity, as it inhibits both BRAF and CRAF. Another approach currently in clinical trials is the antisense oligonucleotide ISIS 5132.128
NF-κB Pathway
The NF-κB pathway plays a pivotal role in cell survival, proliferation, and inflammation by activating transcription of antiapoptotic genes and proinflammatory cytokines.129 Constitutive activation of NF-κB contributes to chemotherapy resistance by increasing expression of multidrug resistance P-glycoproteins. Constitutive activation of the NF-κB pathway can occur in response to oncogenic stimuli, such as Ras-mediated transformation.130 Chromosomal aberrations affecting NF-κB genes are frequent in hematologic malignancies, such as amplification of c-Rel in B cell non-Hodgkin’s lymphoma.131 In chronic myeloid leukemia, the Bcr-Abl fusion protein activates NF-κB by promoting nuclear translocation.132 Activation of IKK by the Tax oncoprotein is important for virally induced transformation of CD4 T cells by HTLV-1.133 Disruption of NF-κB signaling is an important therapeutic strategy in multiple contexts, including viral transformation.
Under basal conditions, NF-κB is maintained in an inactive state and sequestered in the cytosol by binding the inhibitor of NF-κB, IκB. Activating stimuli release NF-κB from inhibition through the phosphorylation of IkB by IkB kinase (IKK). Phosphorylated IkB is then targeted for ubiquit- ination and degradation by the 26S proteasome, which unmasks the nuclear localization signal of NF-κB to allow nuclear translocation and transcriptional activation. An essential activator of this pathway is the IKK enzyme complex that contains catalytic subunits IKKα and IKKβ and the regulatory subunit IKKγ. Knockout studies showed that IKKβ is responsible for IkB degradation in response to environmental stimuli134 and, therefore, represents a potential specific drug target.
NF-KB-Targeted Therapeutics
Large-scale screens by pharmaceutical companies identified several small molecule inhibitors of IKK.135 Although these drugs were shown to induce apoptosis in preclinical studies, many are hampered by a lack of specificity.136 Apart from specific chemical inhibitors, many natural products were found to inhibit the NF-κB pathway. Examples include curcumin, the antioxidant N-acetyl-cysteine, and nonsteroidal anti-inflammatory drugs such as aspirin.137 One mechanism by which such inhibitors may function is by targeting oxidation-sensitive cysteine residues. For example, the cyclopentenone prostaglandin 15d-PGJ2 inhibits the NF-κB pathway by direct covalent modification of a cysteine residue in the kinase activation loop of IKKβ138 or in the DNA-binding subunits of NF-κB, p50, and RelA.139 Treatment with 15d-PGJ2 was shown to inhibit inflammation in several animal models, such as autoimmune encephalitis140 and adjuvant-induced arthritis.141
Adenoviral Gene Therapy to Inhibit Multiple Kinases
Another strategy for targeting survival kinase cascades is gene therapy with the E1A tumor suppressor. The E1A gene was shown to exert antitumor effects through multiple mechanisms, including suppression of RTKs such as Her2/Neu and inhibition of Akt/NFKB signaling. The E1A gene was also proposed to cooperate with chemotherapeutic drugs to induce cell death in otherwiseresistant cell lines, for example, sensitizing hepatocellular carcinoma cells to gemcitabine.142 The mechanism behind this function may involve inhibition of p21 to induce cell death rather than cell cycle arrest following DNA damage.143
TgDCC-E1A is a lipid-based delivery system developed by Targeted Genetics for treatment of solid tumors overexpressing Her2/Neu.144 Preclinical studies of TgDCC-E1A in mouse models of breast and ovarian cancer demonstrated reduced Her2/Neu expression and tumor formation and increased survival. This therapy showed modest tumor regression in Phase I and II clinical trials in patients with breast and ovarian cancer and with head and neck cancer.145
Activation of Survival Kinase Cascades by Ligand Supplementation
Activation of survival kinase signaling is a therapeutic goal for treatment of neurodegenerative diseases. There is strong rationale for therapeutic use of neurotrophins in multiple neurological disorders, such as nerve growth factor (NGF) in the treatment of Alzheimer’s disease and neuropathies of the peripheral nervous system or IGF and ciliary neurotrophic factor in motor neuron atrophy.146 Although the protective effects of growth factor supplementation were proven in various animal models, clinical trials with neurotrophins were disappointing, largely due to poor pharmacokinetics and other problems associated with the use of large polypeptides as drugs. Other methods for delivery of intact neurotrophins were attempted, including gene therapy147 and microencapsulation of genetically engineered cells that secrete neurotrophins.148 However, these strategies are still limited by the ability to cross the blood-brain barrier (BBB) and require surgical intervention.149 Alternatively, the improved delivery of neurotrophins by conjugation to a BBB-delivery vector showed protection in animal models of ischemia.150,151 BBB-permeable small molecule mimetics of neurotrophins are also under development.152
Erythropoietin (EPO) is another cytokine that showed protective effects in animal models of cell injury, including cerebral and cardiac ischemia.153,154 Although mostly known for its roles in regulating hematopoiesis through activation of PI3K/Akt, MAPK, and JAK/STAT signaling pathways, EPO is increasingly being recognized as an important endogenous protective factor produced in the brain following oxidative stresses. Unlike neurotrophins, recombinant human EPO (rhEPO) can cross the BBB and showed significant improvement in early clinical trials with stroke patients when administered intravenously.155 Because prolonged exposure to rhEPO might have deleterious effects on red blood cell mass, other nonerythropoietic variants were proposed to selectively provide neuroprotection.156
Targeting Protein Degradation
The 26S proteasome was implicated in numerous cellular pathways and may be particularly important for the survival of cancer cells.157 Although proteasome inhibition likely impacts many targets, NF-κB activation is tightly regulated by IκB degradation and seems to be highly sensitive to this strategy. The proteasomal inhibitor PS-341 was shown to effectively inhibit NF-κB activity and tumor cell growth in the HTLV-1 Tax transgenic tumor model in vitro, although the correlation in vivo is less consistent.158 PS-341 is currently being used in clinical trials and has shown success in the treatment of hematologic malignancies.159
In addition to its role in the NF-κB pathway, the proteasome controls degradation of the TSC1 and TSC2 gene products after Akt signaling to allow mTOR activation. p53 degradation in MDM2 overexpressing cells also depends on the proteasome, as does caspase degradation after IAP binding, among other targets. Therefore, although proteasome inhibition is a nonspecific strategy, it seems to selectively induce death in cancer cells.
General Inhibitors of Gene Silencing
Epigenetic gene silencing is emerging as an important mechanism by which tumors suppress proapoptotic pathways. Apoptotic elements that are silenced during tumorigenesis include Apaf- 1 in melanomas160 and ARF in multiple types of cancer.161 Another example is promoter methylation of caspase-8 in some childhood neuroblastomas.162 In this situation, reexpression of caspase-8 by gene transfer was shown to sensitize cells to death receptor signaling.163 Alternatively, treatment with DNA methylation inhibitors such as 5-azacytidine164,165 or histone deacetylase inhibitors such as depsipeptide166,167 may rescue expression. As with proteasome inhibition, although strategies to reverse gene silencing are nonspecific, they seem to preferentially impact the survival of cancer cells.
ER Stress and Ca2+ Homeostasis
In addition to the classic intrinsic and extrinsic pathways for apoptosis, the endoplasmic reticulum (ER) is increasingly being recognized as an important organelle in initiating and propagating apoptotic signals.168 Members of Bcl-2 and BH3-only family members are found directly localized to the ER.48 In vivo, ER perturbation occurs in response to viral stress or the accumulation of unfolded proteins. The unfolded protein response is a highly conserved signal transduction pathway that halts protein synthesis and upregulates chaperone proteins and other factors in the secretory pathway in order to allow the ER to regain folding capacity. However, when ER stress exceeds the ability of this program to restore ER function, apoptosis is initiated. Experimentally, excessive ER stress can be induced with pharmacologic agents that prevent N-linked glycosylation, block ER to Golgi transport, impair disulfide bond formation, or disrupt ER Ca2+ stores. After such stimuli, the ER-localized caspase-12 is activated169 and can directly activate caspase-9, independent of Apaf-1 and mitochondrial membrane permeability. The relevance of caspase-12 signaling in humans remains controversial due to reports of truncation polymorphism in humans.170
ER stress leads to release of Ca2+ stores, primarily through the 1,4,5-triphosphate receptor (IP3R) or Ryanodine receptor (RyR) families. This is an important step in amplifying the ER apoptosis signal through the mitochondrial pathway, as disruption of calcium homeostasis can lead to activation of Ca2+-sensitive calpains, calcineurin and Bad activation, and upregulation of factors that induce cytochrome c release, such as the nuclear orphan receptor Nur77/TR3.171 Furthermore, excessive intracellular calcium can directly cause Ca2+ influx to the mitochondria and subsequent membrane swelling and dysfunction.
ER Stress in Neurodegenerative Disorders
Many neurodegenerative diseases are characterized by misfolded protein aggregates, such as hun- tingtin in Huntington’s disease, amyloid- in Alzheimer’s disease, and α-synuclein in Parkinson’s disease. Therapeutic strategies to target this initial stress include peptido-mimetics that block aggregate seeding and fibril formation.172 Although subsequent steps in ER stress signaling can be targeted in vitro, for example, by using Ca2+ chelators, specific drugs targeting this pathway that are applicable to human use are still under development.
Another source of disruption of Ca2+ homeostasis occurs from excessive Ca2+ influx at the plasma membrane. In some neurodegenerative diseases, overactivation of glutamate receptors leads to sustained elevations in calcium, activation of calcineurin and progressive mitochondrial dysfunction, and, ultimately, neuronal degeneration.173 Memantine is an A-methyl-D-aspartate (NMDA) receptor antagonist that prevents this glutamate influx. It was recently approved for treatment of patients with moderate to severe Alzheimer’s disease and showed efficacy as a single agent or in combination with cholinesterase inhibitors.174,175