COMPLICATIONS OF THERAPY WITH NUCLEOSIDE ANALOGUE REVERSE TRANSCRIPTASE INHIBITORS
NRTI drugs were the first to demonstrate clinical efficacy against HIV infection, and they continue to be used in HAART regimens in combination with HIV protease inhibitors (PIs) or nonnucleoside reverse transcriptase inhibitors (NNRTIs).
The long-term use of NRTI drugs was associated with a number of clinically relevant toxicities, including myopathy, hematological toxicities, hyperlac- tatemia and lactic acidosis, neuropathy, pancreatitis, and, more recently, a syndrome of pathological loss of subcutaneous fat tissue (lipoatrophy). Importantly, the toxicity profile of each NRTI drug within this class is unique in terms of the overall risk of long-term complications as well as the tissue specificity of its toxic effects (Figure 23.1). It is also notable that NRTI drugs entered clinical practice at different times (see Figure 23.2), so that some drugs have spanned the “pre-HAART” and “HAART” eras (e.g., zidovudine, didanosine), whereas others were introduced as a component of HAART regimens (e.g., stavudine, lamivudine). There are also newer agents for which clinical experience is limited at present (e.g., abacavir, tenofovir). Hence, the history of NRTI-associated toxicities reflects the availability of specific drugs at the time as well as the influence of other factors that may have been different in the HAART era compared with the period that preceded it.Mechanisms of Action: Therapeutic and Toxic Effects of NRTI Therapy
The therapeutic activity of NRTI drugs is conferred by their ability to inhibit HIV reverse transcriptase, the viral ribonucleic acid (RNA)-dependent deoxyribonucleic acid (DNA) polymerase that allows the virus to create a DNA sequence based on its own RNA template (Figure 23.3). Following intracellular phosphorylation, NRTI drugs compete with naturally occurring nucleotides for utilization by HIV reverse transcriptase.
Incorporation of the “false” (NRTI) substrate then causes DNA chain termination, as these nucleotides lack a hydroxyl group that is critical for chain elongation. The effectiveness of NRTI drugs as inhibitors of HIV replication is, therefore, determined primarily by their ability to inhibit HIV reverse transcriptase. However, the efficiency with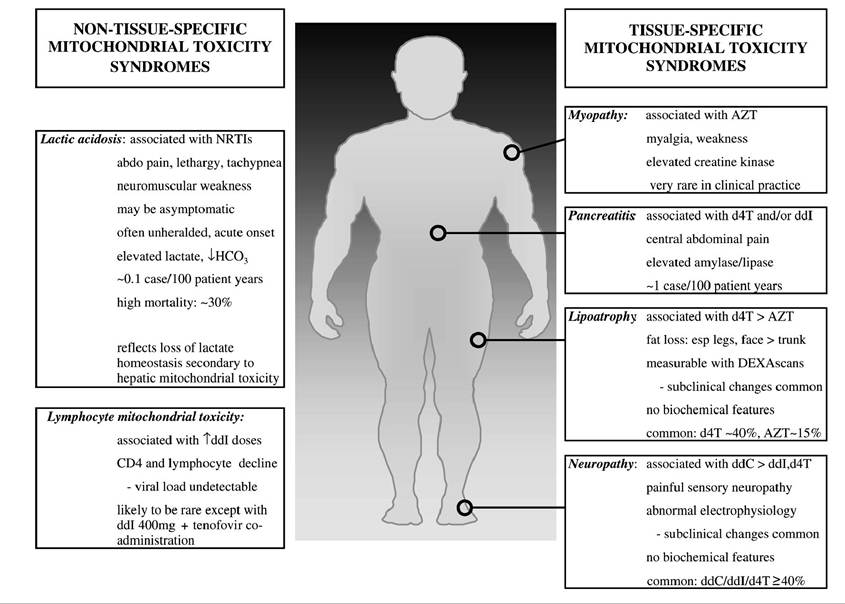
FIGURE 23.1 Clinical syndromes attributed to NRTI-associated mitochondrial toxicity.
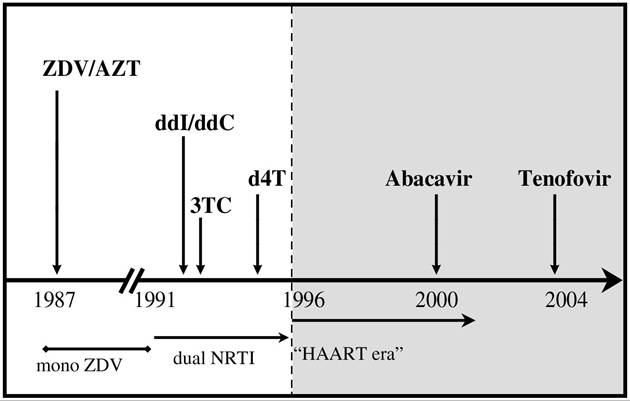
FIGURE 23.2 Time line for the introduction of NRTI drugs into clinical practice in the “pre-HAART” and “HAART” eras.
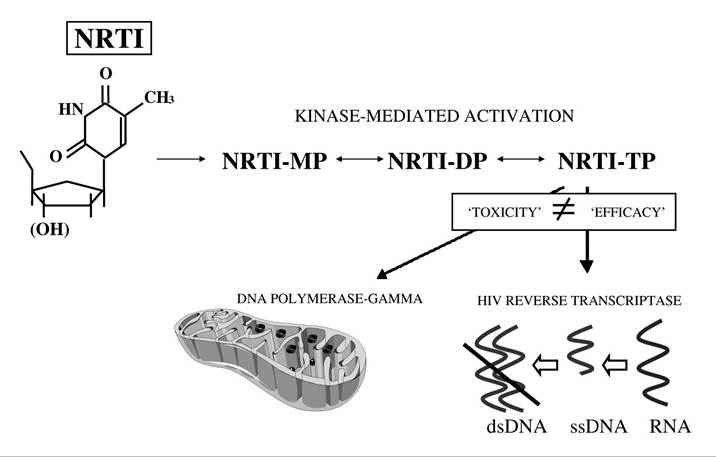
FIGURE 23.3 The basis of NRTI-associated mitochondrial toxicity, according to the “pol-” model. NRTI drugs differ from natural nuclesoide compounds in that the 5-hydroxyl group (OH) is modified or removed, so that incorporation into a nascent DNA chain leads to chain termination. Note that the affinity of a given NRTI drug for DNA polymerase-γ (a determinant of its ability to cause mitochondrial DNA depletion) is not related to its affinity for HIV reverse transcriptase (a determinant of antiretroviral efficacy).
which NRTI drugs are delivered to the appropriate cell population and intracellular compartment— determined by factors such as oral bioavailability, cellular transport mechanisms, and the efficiency of cellular kinases responsible for activating NRTI drugs to their triphosphate form—is also an important factor in determining drug efficacy.
The study of NRTI-associated toxicity syndromes, now more than a decade old, was also informed by their capacity to inhibit a specific host DNA polymerase (mitochondrial DNA poly- merase-γ) through a mechanism that is similar to its therapeutic activity as an HIV reverse transcriptase inhibitor.6-8 However, it is worth stating at the outset that the toxic and therapeutic profiles of individual NRTI drugs are not correlated, so that increased efficacy is not necessarily accompanied by increased toxicity.9 Over the past 15 years, there has been an increasing trend toward the development and clinical use of NRTI drugs (e.g., abacavir, tenofovir) that combine high levels of antiviral efficacy with benign toxicity profiles both in vitro and (thus far) in vivo,8,10 providing cause for optimism that the prevalence of NRTI-associated toxicities will decline over time.
The fact that mitochondria appear to be specifically targeted by NRTI therapy forms the basis for tissue-specific “mitochondrial toxicity” syndromes that may also involve apoptotic cell death as a factor in pathogenesis. As summarized in earlier chapters, mitochondria are both energy powerhouses of the cell and critical regulators of cell death. These organelles are also unique in containing their own genome that is extrachromosomal and replicates independently of nuclear DNA. Hence, mitochondrial DNA can be synthesized in postmitotic and resting cells, using a mitochondrial DNA polymerase (polymerase-γ), with evolutionary origins11 and enzymatic activities12 that are distinct from the multiple DNA polymerases involved in nuclear DNA synthesis. Importantly, DNA pol- is also unable to discriminate strongly in favor of naturally occurring nucleotides (i.e., deoxynucleotides) and against nucleoside analogue drugs,12 so that NRTI drugs are capable of inhibiting cellular mitochondrial DNA synthesis via their effects on DNA pol- (Figure 23.3). The basic premise of the pol- hypothesis is that NRTI-induced inhibition of DNA pol- leads to cellular mitochondrial DNA depletion (i.e., reduced numbers of mitochondrial DNA copies per
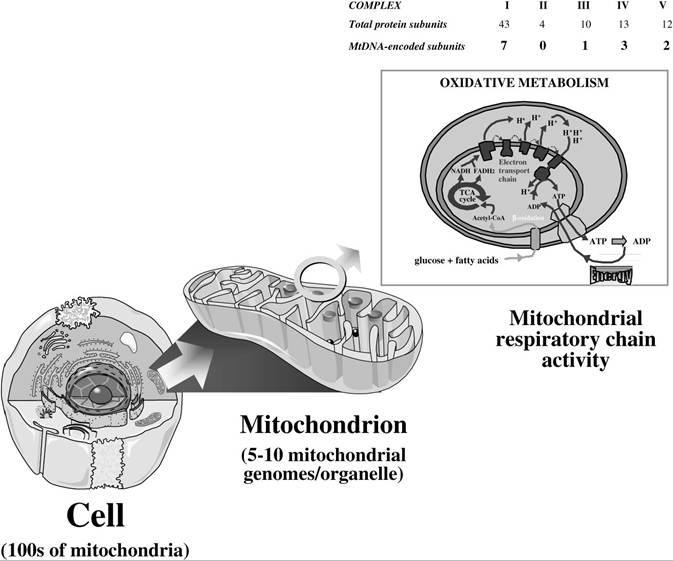
FIGURE 23.4 The mitochondrial organelle and its role in oxidative phosphorylation. Mitochondria are ubiquitous organelles responsible for cellular energy production through oxidative phosphorylation. This requires the concerted action of mitochondrial DNA-encoded subunits (13) and nuclear DNA-encoded subunits (~70) of the five respiratory chain complexes. Note also that all proteins involved in determining mitochondrial organellar number and structure, as well as in maintaining biochemical pathways within the mitochondrial matrix (e.g., oxidation, Krebs’ cycle), are nuclear DNA encoded.
cell). Cellular, and ultimately organ, toxicity is the consequence of loss of mitochondrial bioenergetic function, when mitochondrial DNA levels fall beyond a critical level at which the production of the 13 mitochondrial DNA-encoded protein subunits of the mitochondrial respiratory chain is insufficient to meet energy requirements (see Figure 23.4).
In vitro studies have provided important information regarding the potential of specific NRTI drugs to cause DNA polymerase-γ inhibition, mitochondrial DNA depletion, and mitochondrial dysfunction.8,9,13 A consistent ranking of the potency of mitochondrial DNA synthesis inhibition has emerged from these studies, with decreasing potency associated with ddC (zalcitabine) >> ddI (didanosine) > d4T (stavudine) > AZT (zidovudine). These studies also suggest that no significant mitochondrial polymerase inhibition occurs at pharmacologic concentrations of lamivudine, aba- cavir, and tenofovir. One caveat to these findings is that NRTI-associated mitochondrial toxicity is tissue specific, so that in vitro studies should ideally use human cell cultures that are relevant to the clinical toxicity profile of the drug.
These studies can also shed light on the intracellular activation pathways of specific drugs, which also may influence their ability to affect mitochondria. For example, the thymidine analogues zidovudine and stavudine share a common activation pathway that involves thymidine kinase (TK) in the formation of the monophosphate form of the drug. TK comprises two isoforms: TK1, which is cytosolic and active in mitotically active cells only; and TK2, which is mitochondria specific and active in postmitotic cells. Of interest, stavudine—but not zidovudine—retains anti-HIV activity within the pharmacologic dose range in TK1-deficient cells (IC50 for HIV = 0.27 μM with TK, 2.5 μM in TK- deficient CEM cells)14 and is efficiently phosphorylated to its active triphosphate derivative within isolated mitochondria.15 This suggests that this thymidine analogue is more likely to target mitochondrial DNA, particularly in postmitotic tissues (e.g., nerves) in which mitochondrial DNA synthesis accounts for a greater proportion of nucleoside utilization while nuclear DNA synthesis is latent.
The ability of NRTI drugs to induce cellular mitochondrial toxicity may, therefore, be considered in two parts.
A drug’s affinity for DNA polymerase-γ may be considered a primary determinant, but the relevance of this effect to the development of tissue toxicity is modulated by other factors, such as that drug’s ability to access the mitochondrial compartment within the cell cytosol in an activated form. This is also highlighted by the discovery of a mitochondrial inner membrane transporter that is able to facilitate the entry of active NRTI diphosphate and triphosphate derivatives into mitochondria.16We now move to specific syndromes that have been linked to NRTI treatment in the clinical setting, with a particular emphasis on the distinct toxicity profiles of individual drugs, and the likely role of mitochondrial toxicity and apoptosis in pathogenesis.
Zidovudine Myopathy: A Tale of Two Treatment Eras
Myopathy associated with zidovudine therapy represents the most well-known mitochondrial toxicity syndrome linked to NRTI therapy, as it was the subject of seminal studies and publications at a time when mitochondrial disorders in general were beginning to be appreciated in the late 1980s and early 1990s.17-20 Apoptosis was not directly investigated in these studies—although cell death and tissue destruction were certainly suggested by marked loss of muscle mass in affected areas21—but it was possible to demonstrate abnormal mitochondrial morphology in clinical muscle biopsy specimens, along with significant mitochondrial DNA depletion. Muscle morphology appeared typical of genetically determined mitochondrial myopathy syndromes that were newly described at the time, with evidence of enlarged mitochondria with distorted crystal architecture and the presence of “ragged red” fibers.17-19,21
However, an interesting aspect of this clinical syndrome is its rarity in the current treatment era (~4% in a recent study involving a cohort of 1000 patients)22 compared with previous estimates of ~25%,23 despite the continuous and frequent use of zidovudine throughout the past 17 years.
The most obvious explanation for this dramatic decline is that zidovudine dosing was reduced from 1200 mg (monotherapy) to 500 mg per day (within HAART regimens), although the metabolic activation of zidovudine also appears to be upregulated in poorly controlled and advanced HIV disease, presumably due to increased activity of the rate-limiting TK1 enzyme.24 It also is notable that prolonged and advanced HIV disease is associated with myopathic syndromes and that disease- and treatment-associated pathology may have converged during this period.18,19,21,25 As discussed below, this synergy between HIV and treatment also is observed in the case of neuropathy, although this toxicity syndrome remains prevalent in contemporary practice.Lipoatrophy: Pathological Fat Loss as a Mitochondrial Toxicity
There is now compelling clinical and pathological evidence that the choice and duration of NRTI drugs are the dominant risk factors for developing pathological loss of fat tissue, referred to as subcutaneous fat wasting or lipoatrophy. Moreover, pathogenesis studies have directly implicated apoptotic cell death within adipocyte cell populations as a consequence of NRTI-induced mitochondrial toxicity.
Clinical studies have demonstrated that NRTI therapy alone provides sufficient conditions for the development of lipoatrophy26-28 and that NRTI therapy is an independent risk factor for its occurrence in HAART-treated individuals.29,30 Clinical trials data have now confirmed the findings of observational cohort studies that stavudine therapy is associated with an approximately twofold increased risk of lipoatrophy compared with zidovudine, so that the risk of clinically apparent lipoatrophy over 30 months
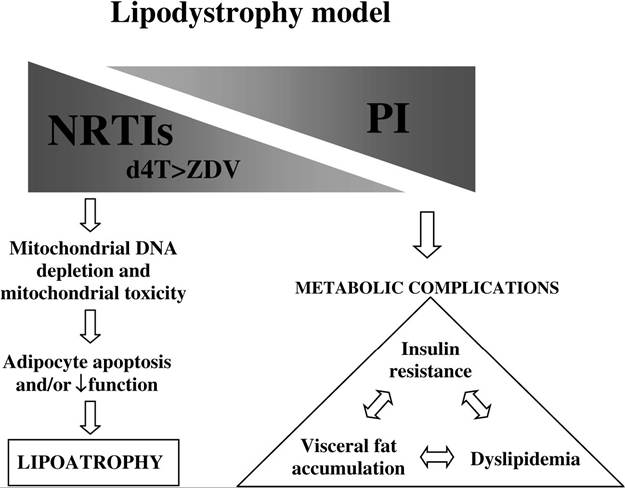
FIGURE 23.5 Model of lipodystrophy pathogenesis, incorporating contributions of NRTI and HIV protease inhibitor therapy.
is approximately 10 to 20% in zidovudine-treated individuals and 40 to 50% among those treated with stavudine.30-32 Switching NRTI therapy from stavudine to abacavir in two clinical trials33’34 was also associated with significant improvements in fat wasting. In contrast, more than 30 trials investigating HIV protease inhibitor discontinuation as a therapeutic strategy (reviewed in Dreschler and Powderly35) failed to demonstrate beneficial effects on fat wasting, although metabolic abnormalities such as dyslipidemia and insulin resistance improved (see Figure 23.5).
The histopathological correlates of this syndrome also indicate that adipose tissue-specific mitochondrial toxicity and widespread adipocyte cell death are central to lipoatrophy pathogenesis. At the ultrastructural level, abnormal mitochondrial architecture and organellar proliferation are prominent features.36-38 Here, the involvement of apoptotic cell death also was characterized, so that increased adipocyte apoptosis associated with lipoatrophy was shown to improve after discontinuing stavudine therapy in favor of abacavir39 but not after switching HIV protease inhibitor therapy.40 In keeping with a role for mitochondrial toxicity in lipoatrophy pathogenesis, studies using precise real-time polymerase chain reaction (PCR) quantitation methods demonstrated significant mitochondrial DNA depletion particularly associated with stavudine compared with zidovu- dine,41,42 and associations were found between NRTI therapy, adipocyte mitochondrial DNA depletion, adipose tissue toxicity, and the eventual development of clinical fat loss.42,43
Neuropathy: Contributions of Both HIV Disease and NRTI Therapy
There are a number of issues to be considered in addressing potential associations between NRTI therapy, mitochondrial toxicity, and neuropathy. First, routine clinical assessment of potential neuropathic changes—relying on the assessment of neurological function rather than more visible changes such as lipoatrophy—may underestimate the prevalence and potential long-term impact of neuropathy in HIV-infected patients. Second, there is a definite contribution of HIV disease per se to the pathogenesis of a form of distal sensory neuropathy that is clinically and electrophysiologically indistinguishable from so-called “toxic neuropathy from antiretroviral drugs” (reviewed in Keswani et al.44). It is, therefore, difficult to dissect the relative contribution of disease-associated and drug- associated factors in the syndrome, as these effects are likely to be synergistic. The clinical syndrome common to both HIV-associated and toxic sensory polyneuropathy is dominated by peripheral pain and dysesthesia, with rare motor involvement.
In general, the relative risk of toxic polyneuropathy associated with specific NRTI drugs correlates well with their ability to inhibit mitochondrial DNA polymerase-γ in vitro, with greatest risk associated with zalcitabine, with equivalent affinity for mitochondrial DNA polymerase-γ and HIV reverse transcriptase that provides for a narrow therapeutic window.9 Neuropathy was a consistent and dose-limiting toxicity in trials of zalcitabine, occurring in approximately one-third of patients receiving low-dose therapy (2.25 mg/day) for less than a year,45,46 whereas higher doses were associated with almost universal development of neuropathy (100% with >0.04 mg/kg/day, 80% with 0.04 mg/kg/day).45 Consistent with the pol- hypothesis, Dalakas and colleagues47 provided in vivo evidence of significant mitochondrial DNA depletion in peripheral nerves associated with this complication of zalcitabine therapy, although studies investigating potential roles of apoptosis were not published to date.
Stavudine and didanosine also were associated with increased risk of neuropathy in the large Johns Hopkins AIDS Service cohort study (n = 1116). Compared with a crude incidence rate of 6.8 cases per 100 person years for didanosine, stavudine use was associated with a relative risk of 1.4, and concurrent use of these drugs further increased risk (3.5-fold relative risk for stavudine/ didanosine compared with didanosine alone). Concurrent hydroxyurea therapy had an additional effect (7.8-fold relative risk of stavudine/didanosine/hydroxyurea compared with didanosine alone, giving a crude incidence of 28.6 cases per 100 person years).48
As already mentioned, markers of progressive HIV disease correlate with the development of sensory polyneuropathy, with evidence for associations between reduced CD4+ T cell counts and increased risk of neuropathy (reviewed in Weissman49), and increased pain scores in patients with higher HIV viral load.50 However, in a cohort of 144 ambulatory patients with longer duration of exposure to stavudine or didanosine and average CD4+ T cell counts >400 ? 106/L, the prevalence of sensory neuropathy was found to be 44%,51 suggesting that among patients with prolonged survival and effective immunologic and virological outcomes, choice and duration of NRTI therapy will ultimately become the dominant risk factors for neuropathy. In this analysis, use of stavudine, didanosine, or zalcitabine compared with other NRTI drugs was associated with an odds ratio of 26 of developing clinical neuropathy, whereas disease-associated factors were not found to be significant in multivariate analysis.
"Lymphocyte Mitochondrial Toxicity" and Didanosine Therapy: An
Intersection of Therapeutic and Toxic Effects
Intriguing results from a recent study has suggested that a suboptimal immunologic response to antiretroviral therapy (i.e., loss of CD4+ T cell counts on treatment) may represent a complication of NRTI therapy in selected circumstances.52 These data emerged from a Spanish cohort study involving 302 patients, in which regimens containing standard doses of didanosine (ddI; 400 mg) and tenofovir were associated with significant decreases in CD4+ and CD8+ T cells as well as total lymphocyte counts (with a decline of >100 CD4+ T cells at 48 weeks in more than 50%). Plasma levels of ddI appeared to be pharmacologically “boosted” in all patients receiving coadministered ddI 400 mg and tenofovir and returned to expected levels when the ddI dose was reduced to 250 mg.52 More recently, a case series was reported of four patients who developed an identical clinical syndrome while receiving zidovudine and ddI with a mean reduction of CD4+ lymphocyte count from 442 to 220 cells/pL,53 when switching from ddI to lamivudine proved to be an effective strategy (mean CD4+ lymphocytes increased to 385 cells/pL).53
This is not to suggest that immunologic responses to ddI are generally inferior to other NRTI drugs, as this drug has demonstrated efficacy in large clinical trials.54’55 However, this emerging field of investigation provides an interesting insight into the tissue-specific nature of NRTI toxicity and the potential for drug toxicity to directly impinge on the efficacy of HIV treatment. Associations between ddI therapy and peripheral blood mitochondrial DNA depletion have been noted by a number of groups,41,56-58 whereas this drug has not been associated with any significant effect on mitochondrial DNA in subcutaneous fat samples obtained concurrently with blood samples.41 Hence, there is a sound basis for considering a potential role for ddI toxicity when “discordant” responses to treatment are observed. Other chapters addressed the critical role of lymphocyte apoptosis in determining T cell dynamics both before and after treatment, placing these data in a broader context.
Lactic Acidosis and Hyperlactatemia
Lactic acidosis is probably the most recognizable feature of mitochondrial toxicity in clinical disease, in which loss of mitochondrial oxidative function leads to increased reliance on “anaerobic” metabolism and the inevitable accumulation of lactate (and thus, of acid). In the setting of NRTI therapy, there is now an appreciation of a spectrum of clinical phenotypes associated with elevated systemic lactate levels.59 At one end of this spectrum is a relatively common syndrome of mild, asymptomatic, nonprogressive hyperlactatemia (generally < 2.5 mmol/L), which appears to represent a “compensated” homeostatic system in which elevated lactate production is balanced by effective mechanisms of lactate clearance. Although the degree of hyperlactatemia seems to be greater in the presence of stavudine or ddI therapy than in the presence of zidovudine or abacavir, this syndrome appears to be benign, irrespective of the choice of NRTI therapy.60-64
In contrast, lactic acidosis and hepatic steatosis are uncommon (approximately 1 to 2 per 1000 person years) but carry a high mortality rate (~50%),65 representing a syndrome of “decompensated” lactate metabolism that allows the rapid, progressive accumulation of lactate and development of acidosis in affected patients. A critical aspect of lactic acidosis is its unpredictability, as it typically occurs in patients who were on stable NRTI regimens for months or even years, and it is not heralded by increased lactate levels before the development of the fulminant syndrome. Host risk factors have been identified for NRTI-associated lactic acidosis, including concurrent liver disease, female gender, and obesity. Although the majority of reported cases in recent years have involved stavudine-based HAART, it is important to recognize that cases involving zidovudine therapy also occur (reviewed in John and Mallal59). A more recently identified clinical syndrome accompanying lactic acidosis is progressive, severe neuromuscular weakness mimicking the Guillain-Barre syndrome.66 Initial reports by the Food and Drug Administration identified 25 cases, including seven fatal cases (notably, NRTI therapy was continued after the diagnosis was made in all but one of these individuals). A more recent retrospective analysis identified 55 cases (44 associated with stavudine therapy), with a 25% mortality rate among the 36 patients with documented follow-up.67
With regard to the pathogenesis of hyperlactatemia syndromes, evidence has been provided from studies using exogenous lactate loading that show that increased lactate production is the fundamental defect in chronic asymptomatic hyperlactatemia rather than decreased clearance of lactate (which primarily involves the liver).68 Lactate clearance seems to be upregulated as a compensatory response to an elevated systemic lactate load.68 In contrast, lactic acidosis represents a complete failure of lactate homeostasis, in which lactic acid accumulation progresses inexorably while NRTI therapy is maintained. In this scenario, the prominent role of liver mitochondrial dysfunction (indicated by the presence of marked microvesicular hepatic steatosis),69,70 leading to a shift from hepatic lactate consumption to lactate production, would be predicted to have dramatic consequences for systemic lactate metabolism. The capacity for selected NRTI drugs (zalcitabine, ddI, and stavudine) to induce significant mitochondrial DNA depletion in liver biopsy samples71 also lends support to the possibility that functional hepatic reserve may be impaired and predisposition to hepatic steatosis (reflecting mitochondrial dysfunction and impaired hepatic β-oxidation) may be increased.