HOST GENETIC FACTORS THAT INFLUENCE DISEASE PROGRESSION
There are several host genetic factors that are postulated to influence viral load set point and rate of disease progression. Host-specific alterations in soluble mediators or their receptors, host human leukocyte antigen (HLA)-type, and gender are genetic factors presumably involved in influencing the rate of disease progression.
Chemokine Receptor Mutations
After identification of HIV as the causative agent of AIDS, it became evident that the presence of CD4 molecules on target cell surfaces was necessary but not sufficient by itself for viral entry, raising the possibility that a second receptor may be needed for HIV to enter a cell. The identification of HIV-inhibitory chemokines, such as regulated upon activation normal T cell expressed and secreted (RANTES), macrophage inflammatory protein (MIP)-∣α, MIP-1β, and stromal derived factor (SDF)-1, as well as their natural receptors CCR5 and CXCR4, led to the observation that these chemokine receptors served an important co-receptor function for HIV that allowed infection of CD4+ cells.13-15
Investigators identified two men who were exposed to HIV on repeated occasions but remained uninfected. Examination of these two subjects led to the identification of a 32-base pair deletion in the CCR5 chemokine receptor referred to as the CCR5-∆32 mutation that explained their resistance to infection by certain strains of HIV. The two subjects were found to be homozygotes for this mutation, resulting in alteration of CCR5 such that viruses using this co-receptor could not infect CD4+ cells.16 A subsequent study of 1252 men who have sex with men (MSM) enrolled in the Multicenter AIDS Cohort Study (MACS) revealed that the mutant allele had a relatively high frequency in the general Caucasian population (0.08%) but was comparatively overrepresented in the at-risk, HIV-uninfected MSM population—3.6% of them were homozygotes.
In combination with the observation that none of the HIV-infected MSM were homozygotes, CCR5-∆32 homozygosity was postulated to be protective against strains of HIV infection requiring CCR5 for a coreceptor (R5 strain).17 Cases of HIV infection in individuals homozygous at this allele are caused by viruses that use receptors other than CCR5, such as CXCR4, CCR2, and CCR3.18Heterozygotes with the CCR5-∆32 mutation also seem to have a slower rate of disease progression, despite only being partially protected against infection with the R5-strain HIV.19 As expected, the protection afforded by heterozygosity for CCR5-∆32 is lost if the infecting virus uses CXCR4 (X4 strain) as its co-receptor.20 It is important to note that there are individuals who are highly exposed to HIV and remain uninfected who do not harbor any known co-receptor polymorphisms. The operative mechanism providing protection from infection in these cases is unknown.
Mutations in SDF-1 (the natural ligand of CXCR4), specific polymorphisms of CCR5, and homozygosity for a specific haplotype of a newly described receptor CX3CR1 are additional putative host genetic characteristics that may influence the immunologic and virologic course of HIV infection.21
Conversely, differential expression of certain proinflammatory cytokines such as tumor necrosis factor (TNF)α or interleukin (IL)-1β, as well as nonspecific immune activation by other infections such as helminths, seems to correlate with enhanced replication and higher viral loads in infected individuals.22,23 Interestingly, treatment of helminthic infections in a cohort of patients in Ethiopia resulted in a significant decrease in viral load, although there was no difference in initial viral load between helminth-infected and helminth-free subjects.24
Human Leukocyte Antigen (HLA)
The HLA genes are located on the short arm of chromosome 6 and make up the human major histocompatibility complex (MHC).
The two classes of MHC are coded by HLA class I and II genes. HLA-A, HLA-B, and HLA-C encode the MHC class I molecules expressed on all nucleated cells that are responsible for presenting antigen to CD8+ cytotoxic T cells. HLA-DR, HLA-DQ, and HLA-DP encode the MHC class II molecules expressed on the surface of antigen-presenting cells that are responsible for displaying antigen to CD4+ helper T cells. These six HLA loci are considered the most polymorphic in the human genome, allowing humans to respond to multiple and diverse infectious challenges.25 Because the cellular immune system relies on presentation of viral antigen within the context of HLA molecules, it is conceivable that HLA molecules and their interactions with viral peptides would influence the efficiency and effectiveness of the cellular immune response.Several associations have been made between HLA genotype and HIV disease progression and susceptibility. Certain class I alleles, such as B57, B27, and B35, are associated with variable rates of disease progression. Additionally, increasing heterozygosity at the three HLA class I loci, irrespective of specific allele, seems to confer some level of protection from disease progression.25 This observation is thought to confirm the theory of overdominant selection of MHC molecules. This hypothesis posits that HLA heterozygotes have more combinations of MHC molecules and, therefore, have an enhanced ability to present a greater diversity of epitopes than homozygotes. Therefore, diversity at HLA alleles may be expressed phenotypically by a broadened ability to respond to varying pathogens, conferring a selective advantage.26 In one study, 498 individuals from five AIDS cohorts were recruited and HLA typed. Those homozygous at one HLA class I locus progressed to AIDS more rapidly than heterozygotes. More loci for which individuals were homozygous resulted in an outcome that was worse. Heterozygotes fared better than single-locus homozygotes, who, in turn, fared better than individuals homozygous at two or three of their HLA loci.26 Similar data were obtained in Dutch and Rwandan cohorts.27
Though diversity seems important in immunologic control of HIV infection, specific alleles have also been characterized as being protective or deleterious in HIV progression.
The three best- studied HLA class I alleles are HLA-B57, HLA-B27, and HLA-B35. Less is known about specific HLA II alleles.Both HLA-B57 and HLA-B27 are associated with slower disease progression. One hypothesis is that HLA-B57 is able to present multiple HIV epitopes, providing a broad response as well as limiting the possibility of viral escape.25 HLA-B27 seems to recognize and present a HIV-1 Gag epitope that is under evolutionary pressure to remain stable, limiting variation of that epitope and eventual immunologic escape.25 It seems, however, that whole alleles may not be the only prognosticators of disease progression and immune control.
HLA-B alleles may be divided into two groups based on differential amino acid sequences at the carboxyl ends of their αl helices. The Bw4 group includes HLA-B57, HLA-B27, and several other loci. The Bw6 group includes, among others, HLA-B35. Individuals heterozygous for an HLA-B allele may still be considered homozygous for their Bw group. For instance, a heterozygote expressing HLA-B57 and HLA-B27 has two Bw4 alleles, making them Bw4 homozygotes. One study compared HLA types of 39 HIV-positive individuals, 20 of whom were characterized as “controllers of viremia.” Analysis revealed that Bw4 epitope group homozygosity was associated with control of viremia and slowed disease progression, whereas the presence of a Bw6 allele, such as HLA-B35, was not.28 Interestingly, epitopes presented in the context of the Bw4 motif are involved in blocking natural killer cell inhibition, whereas peptides presented in the context of the Bw6 motif are not. This observation led to the conclusion that all HLA homozygosity may not be deleterious in the immune handling of HIV.28
The Bw6 allele HLA-B35 is associated with increased susceptibility to progression of HIV infection to AIDS. Differential rates of progression have been noted in Caucasian and African- American patients who carried the HLA-B35 allele.
In one study, it seemed that HLA-B35 was correlated with worse outcome in both groups, but more so in Caucasians.26 Further study revealed that the HLA-B35 allele could be divided into subtypes HLAB35-Px and HLAB35-Py based on peptide specificity at epitope positions 2 and 9. Analysis of a cohort of Caucasian and African-American patients revealed that the HLA-B35-Px subtype was associated with faster disease progression, whereas the HLA-B35-Py was not. The lower frequency of HLAB35Px subtype alleles in association with the higher occurrence of HLAB35Py alleles in the African-Americans studied was postulated to explain the previous racial differences in disease progression noted among individuals who carried the HLAB35 allele.29 Interestingly, the same study demonstrated that the HLA-Cw4, previously considered an allele that portended rapid progression to HIV, did not do so independently of HLA-B35-Px. Its postulated role as a marker of rapid disease progression is likely attributed to linkage.29Significantly fewer durable associations have been made between disease progression and HLA- II alleles. HLADRB1*13 is frequently cited with mixed disease progression outcomes, some due to race, raising the possibility of further complexity of this allele and its role in HIV disease progression.25 One study noted better virologic outcome with highly active antiretroviral therapy (HAART) in subjects with the HLA-DR1*13 haplotype. This improved outcome was attributed to optimized MHCII HIV epitope presentation, leading to a more effective anti-HIV Th2 effector response as demonstrated by lymphocyte proliferation and interferon-γ secretion.30 Further scrutiny of HLA II alleles will be necessary to better understand their role in progression of asymptomatic HIV infection to AIDS.
Gender
Gender seems to be another genetic factor that may impact viral load set point and, ultimately, disease progression. Several studies have been published comparing differences in viremia between male and female subjects infected with HIV.
A meta-analysis of these studies revealed that women have a 41% lower viral load set point yet progress at the same rate for a given CD4 count and disease stage than men. This observation raises the possibility that women may progress more rapidly than men to AIDS at a given viral load set point. Women, therefore, may need to be treated more aggressively given the gender-specific magnification of the viral load’s impact on progression to AIDS.31 The mechanism of this phenomenon is not known, and no specific guidelines exist for differential interpretation of viral load in men and women.immunologic factors impacting disease progression
Unlike the Herpesviridae family, HIV produces chronic active, rather than latent, infection. Several observations regarding HIV and the immune response in infected subjects provide insight into the lack of persistent immune control of HIV. A group of HIV-infected subjects was described with members who have vigorous HIV-specific immune responses and spontaneously control viremia in the absence of antiviral therapy. Many of these patients harbor robust cytotoxic and T helper cell responses, a finding that implies that for some, cellular immunity may limit HIV to a latent infection. These patients are the exception, however, rather than the rule.
The vast majority of HIV-infected individuals only partially contain viral replication and often progress to AIDS. Although the precise mechanisms for immunologic success or failure remain to be elucidated, there are several observations that shed insight into HIV pathogenesis. Impairment of HIV-specific T helper cell function is one of the most notable defects in the immunologic repertoire of infected hosts. Early in HIV infection, the initial viremia triggers a robust immune response that leads to the activation of as many as 10% of CD8+, cytotoxic T lymphocytes (CTLs).32 These cytotoxic T cells are postulated to be the mechanism behind the initial decline in viremia and establishment of a viral load set point. After the initial viremia begins to decline, HIV-specific neutralizing antibody becomes detectable, lending supportive evidence for the primary role of CTLs rather than antibodies in viral containment. After 6 to 12 months of infection, a viral set point is reached that influences the rate of disease progression. Higher viral loads are correlated with more rapid decline in CD4 cell count and more rapid disease progression.
Individuals with long-term nonprogressing infection and chronically infected persons with undetectable plasma viral loads on therapy harbor latent HIV provirus in their lymph nodes and spleen.33 Mathematical models estimate that a 60- to 73-year course of therapy would be necessary to eradicate the latent reservoir of HIV, suggesting that the eradication of HIV with potent antiviral therapy is essentially unachievable.33 Some evidence exists that early initiation of antiretroviral therapy in the setting of recent infection may lower levels of latent viral reservoirs.34
Three key immune responses are postulated to be important in control of HIV infection. These are neutralizing antibodies, CD8+ cytotoxic T lymphocytes, and CD4+ T helper cells. Impairment of HIV-specific T helper cell responses in chronic infection is likely to cause a significant immunologic defect in both humoral and cellular immune responses. First, we will discuss the role of HIV-specific neutralizing antibodies and cytotoxic T cells. Second, we will review the role of CD4+ T helper cells in HIV-specific immunity and discuss potential mechanisms involved in the success or failure of immunologic control of HIV infection.
Antibody Response
Within 1 to 3 months after infection, HIV antibodies are detected. Nonneutralizing antibody production begins to appear 2 to 3 weeks after infection, although these antibodies do not seem to play a role in the initial decline in viremia.35 These antibodies are directed against HIV-1 structural proteins such as p24 and p17 from Gag. Subsequently, antibodies against HIV-1 Env and Pol as well as other accessory and regulatory proteins develop over time.36 It is not well understood, however, what role these early antibodies play in the overall immunologic control of HIV infection, because they may not target neutralizing epitopes. Neutralizing antibodies targeting the viral envelope are formed in early infection and may be able to bind HIV before entry into target cells. One potential limitation of detected antibodies is that they may be directed against viral debris rather than true functional epitopes.37-39 The role of neutralizing antibody in control of viremia remains unclear. For instance, some studies indicate that the presence of neutralizing maternal antibodies may decrease perinatal HIV infection.40 Other studies, however, provide conflicting evidence that the presence of neutralizing maternal antibody does not protect from transmission.41 Another study of health care workers exposed to HIV-positive blood through needlestick injury correlated the presence of early neutralizing antibody with decreased viral load compared with workers who did not develop neutralizing antibody.42 In another study, some subjects treated during primary HIV who subsequently underwent treatment interruption seemed to develop neutralizing antibody responses temporally associated with a decline in viremia after viral rebound.43 The role of HIV-specific antibody responses in immunologic control of HIV infection is further complicated by three observations:
1. Some HIV-infected individuals with long-term nonprogressive infection maintain strong neutralizing antibody responses, whereas others do not.44
2. Viremia during acute HIV infection declines before neutralizing antibodies are detectable, providing evidence that the neutralizing antibody response may not be the primary mechanism in controlling viral replication.45
3. Neutralizing antibodies through passive immunization protects monkeys from challenge infection with laboratory strains of virus, but high titers of these antibodies are required to prevent infection by primary isolates from humans or monkeys not passed through cell culture.46
Though not all antibodies are neutralizing, they may serve other functions in controlling HIV, such as complement-directed lysis of infected cells as well as antibody-dependent cell-mediated cytotoxicity (ADCC).47
HIV may evade an antibody response by several mechanisms. Given the poor proofreading mechanisms of HIV, rapid viral diversification may result in escape from neutralizing antibody. This antibody escape was observed in a laboratory worker infected with a strain of virus known to be susceptible to neutralizing antibody. The infecting virus ultimately developed variants that were resistant to antibody that previously had been neutralizing.48 Another mechanism postulated to be involved in escape from neutralization is glycosylation. Subjects studied after acute seroconversion developed successive strains of neutralization-resistant virus.49 Sequence analysis revealed very few mutations of the Env gene. Additionally, these mutations were not in areas that coded sequences known to be important targets of HIV-neutralizing antibody. It was observed that increased glycosylation developed on these neutralization-resistant viruses, raising the possibility that they develop a “glycan shield” that sterically inhibits neutralizing antibody from binding to virus.49 Steric hindrance of antibody attachment by overlapping hypervariable loops may also be responsible for failure of neutralization.50 Neutralizing antibody continues to be the subject of intense research scrutiny given its possible application in HIV vaccine development.
Cytotoxic T Lymphocytes
The first effector cell to play a role in viral containment is the CD8+ cytotoxic T lymphocyte. Initial viremia triggers a robust immune response that leads to the activation of as many as 10% of the infected host’s CD8+, cytotoxic T lymphoctes.32 This response is temporally associated with the decline of viremia observed during primary HIV. Multiple lines of evidence in both humans and monkeys indicate the importance of this response in the immunologic control of HIV. Experimental depletion of CD8+ cells in SIV-infected monkeys results in a rebound in viremia. As CD8+ cells are replenished, viremia declines. If CD8 depletion is sustained, so, in turn, is the increase in the SIV load.51
Tetramer studies confirm that cytotoxic T lymphocyte activity coincides with peak viremia.32 Peak in HIV-specific CTLs occurs before the development of neutralizing antibodies, supporting the role of virus-specific CTLs in control of HIV. Initially, the CTL response is limited to a few epitopes in most acutely infected individuals. This narrowly directed response may reflect the fact that the virus is relatively homogenous in early infection. Some theorize that the diversity in CTL response is important prognostically. HIV-infected individuals may generate a very narrow CTL response to HIV in acute infection that is associated with poor immune control of the virus. This narrow response may lead to overexpansion of the CTL clones and immunologic exhaustion.52
HIV-specific CTLs in chronic stages of infection persist over time. Tetramer staining reveals that even in later stages of infection, 0.1 to 2% of circulating CD8 CTLs are HIV specific.53 The persistence of the HIV-specific CTL response seems to require ongoing stimulation with HIV antigens. Tetramer staining of lymphocytes in patients on antiretroviral therapy reveals a steady decline in the number of circulating HIV-specific CTLs.54'55 In untreated disease, the high number of HIV-specific CTLs persists as disease progression to AIDS occurs. The precise mechanisms of failure of cytotoxic T cell immunologic control of HIV are not well understood.
T Helper Cells
CD4+ T helper (Th) cells are critical in coordinating and maintaining host immune responses to viral infection. One role of CD4+ cells is to prime cellular immunity. A major function of Th cells is to stimulate antigen-presenting cells (APCs), leading to the activation of CD8+ effector cells. In a murine model of cytotoxic T cell activation, CD4+ lymphocytes were necessary to activate the antigen-presenting dendritic cells of immunized mice, ultimately allowing MHC I restricted CD8+ cells to kill cells presenting foreign antigen on their surfaces. In the absence of priming of APCs by epitope-specific CD4+ cells, surrogate anti-CD40 antibody acting in lieu of the CD4+ CD40+ cells, or helper-independent viral infection of the dendritic cell, the CD8 effector response was ineffective.56 One can postulate, therefore, that the loss of Th activity through direct depletion or lack of appropriate activation would result in a suboptimal CD8+ response and ineffective clearance of virus.
In addition to priming, Th cell function has been postulated, through in vitro and in vivo evidence, to be necessary to maintain robust virus-specific cytotoxic T cell responses necessary to contain viral replication. One example of the dependence of CTL on CD4+ T cell help was observed in human cytomegalovirus (CMV) infection. Clones of CMV-specific CTLs were adoptively transferred into bone marrow transplant patients at high risk for CMV infection during the course of their procedure. The cytotoxic T cell activity was maintained in subjects with sufficient CMV- specific Th cell responses but rapidly waned in persons without adequate virus-specific Th cell activity. The level of cytotoxic activity waned at a much faster rate in recipients who did not demonstrate a CMV-specific CD4+ response than in those patients who did.57 Adoptive transfer of HIV-specific CTL similarly resulted in waning of activity, presumably due to the lack of appropriate CD4+ T cell help needed to maintain their response.58
Adoptive transfer of HIV-specific CTL accompanied by large doses of IL-2 resulted in maintenance of the HIV-specific CD8+ response. This cytokine infusion was postulated to provide “help” to the transferred CTL via an IL-2 dependent pathway, potentially mimicking the activity of T helper cells. This population of CTL effectively cleared viremia and, by so doing, was thought to have exerted an evolutionary pressure on HIV. Ultimately, sequence variants were generated that escaped the clonal specificity of the infused cytotoxic lymphocytes, resulting in recurrent viremia with a mutant virus.59
Additional support for the role of CD4+ T helper cells in generating and maintaining cellular immune responses is provided by a murine model of viral infection with lymphocytic choriomeningitis virus (LCMV).60 In this model, CD4+ knockout mice were compared to their CD4+ wildtype counterparts. When infected with the indolent Armstrong strain of LCMV, both the knockout and wild-type mice produced a vigorous CD8+ CTL response at the onset of infection, achieving clearance of the LCMV viremia. When the same populations of mice were exposed to more virulent LCMV strains, several differences in the responses were observed. Both strains of mice were able to produce CD8+ cells, but the CD4+ knockout mice had waning virus-specific CTL responses and eventually lost control of viral replication. Antibody-mediated depletion of CD4+ cells in the wildtype mice yielded results similar to those noted in the CD4+ knockouts. Replenishing the depleted CD4+ cells in these mice did not result in a “wild-type” LCMV-specific immune response.60 Similar data in a murine model of Epstein-Barr virus (EBV) infection also demonstrated that CD4+ cell activity is likely involved in maintaining CD8+ effector cell control of viremia.61
Extrapolating from these models, CD4+ Th responses also seem to be important in the control of HIV replication and disease pathogenesis. An inverse correlation between HIV-specific CD4+ Th response and viremia in chronic infection has been demonstrated.62 Similarly, some persons with long-term nonprogressive infection who are able to spontaneously control viremia in the absence of antiretroviral therapy typically have robust CD4+ and CD8+ responses. Additionally, in some subjects treated during acute infection who generated and maintained an HIV-specific T cell response, viremia was transiently controlled even when therapy was interrupted. Similar to the LCMV murine model described above, in vitro depletion of CD4+ cells in the PBMC of a patient with known CTL responses against HIV epitopes results in loss of these responses.62,63 Murine models of retrovirus infection with Friend virus (FV) also seem to indicate that the CD4+ compartment is necessary in maintaining control of viral infection. Depletion of CD4+ cells in these models resulted in FV replication, viral activation from reservoir B cells, as well as signs and symptoms of active disease.64
In HIV-infected individuals, virus-specific cellular immune responses seem to be maintained by treatment with HAART early in primary infection.65 Comparisons of subjects initiated during early or later phases of infection demonstrate differences in in vitro HIV-specific immune responses. In chronic HIV infection, virus-specific proliferation is typically weak or absent. Despite effective therapy with HAART, most chronically infected individuals do not restore HIV-specific proliferative responses. In persons treated during acute HIV infection, subjects maintained on HAART had persistent HIV-specific Th responses as well as persistent CTL activity. Similarly, subjects treated
early, but in an interrupted fashion, displayed durability of their HIV-specific CD4+ and CD8+ proliferative responses.66 Transient immunologic and virologic control in HIV-infected individuals was also reported in the context of supervised treatment interruptions (STI) of HAART in small cohorts treated early in the course of acute/early HIV infection. Both CD4+ and CD8+ responses are preserved in this group, with some evidence that the immunologic repertoire may become more diverse.65
Chronically infected individuals not treated during early infection lack HIV-specific CD4+ proliferative responses. More recently, evidence has emerged that the lack of CD4 proliferation does not correlate with an absolute absence of HIV-specific CD4 cells or Th1 cytokine production in chronically infected patients.67 Furthermore, there is a dichotomous response observed in which there is cytokine secretion but no proliferation.68 A recent study demonstrated that these cells are present in chronically infected individuals and are able to produce intracellular cytokines such as IFN- but do not proliferate in the presence of HIV-specific CD4 antigens.68 The lack of proliferation implies that the response exists in these individuals but that it may somehow be impaired or down- regulated.
CD4+ T Cell Impairment
A defect of the HIV-specific CD4+ response results in significant immunologic impairment. One hypothesis is that during primary HIV infection, virus-specific T helper cells are activated and made more susceptible to infection and HIV-mediated cell death.69
Several mechanisms have been postulated to be involved in the impairment of CD4+ T helper responses. All of these pathways to CD4 attenuation have been characterized through in vitro evidence, so the significance of these pathways is unclear in the infected host. These theories include the following: apoptosis, HIV-mediated cell killing, syncytia formation, antibody-dependent cellular cytotoxicity, and deletion by HIV-specific cytolytic T lymphocytes (Figure 12.1).70
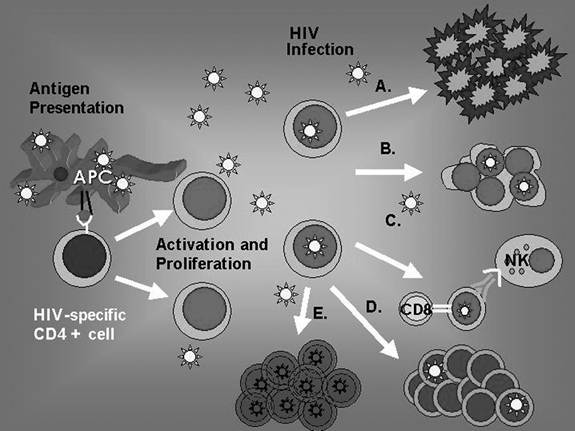
FIGURE 12.1 Antigen is presented to HIV-specific CD4+ cells causing them to activate and proliferate. Infection of CD4 cells with HIV is postulated to trigger several pathways involved in attenuation of the HIV- specific CD4 response. These include direct HIV-induced cytotoxicity and apoptosis (A.), syncytia formation (B.), cell-mediated cytotoxicity by means of CD8+ CTL and natural killer cells (C.), and HIV-induced CD4 dysfunction.70 Additionally, some infected cells that do not die revert to resting CD4+ cells and reservoirs for latent HIV (E.).
Apoptosis
Apoptosis is one mechanism postulated in the HIV-mediated impairment of HIV-specific T helper cells. Several mechanisms are thought to be involved in HIV-induced apoptosis of effector cells. Apoptosis involves several classes of proteins that regulate the process of programmed cell death. The proteins that are involved in initiating apoptosis are cysteine-dependent aspartate-specific proteases or caspases. When signaled to undergo programmed death, the cell’s caspases are triggered, changing the activation state of multiple intracellular proteins to result in mitochondrial, nuclear, and membrane changes characteristic of apoptosis. Terminal commitment to apoptosis is characterized by activation of caspase-3.71
HIV is postulated to enhance programmed cell death in both infected and uninfected cells through several pathways. Among these mechanisms, CD4+ T lymphocytes in HIV-infected subjects express higher levels of Fas and Fas ligand than similar cells in uninfected controls, and the number of the Fas-enhanced cells seems to increase in later stages of infection.72-73 One potential mechanism for increased Fas/Fas ligand expression is thought to be induction by the HIV proteins Nef, Env, and Tat. HIV proteins may also alter the balance of regulatory proteins favoring programmed cell death in the cells it infects.71 It is not, however, optimal for HIV’s survival to immediately induce apoptosis in infected cells. Infection with the virus shortens the life of the CD4 cell and promotes apoptosis, but it seems as if uninfected bystander cells may undergo apoptosis more readily than their infected counterparts.74,75 Uninfected cells are thought to undergo apoptosis due to HIV proteins released from infected cells or by activation-induced cell death.71
Several soluble HIV proteins have been found to cause uninfected cells to undergo programmed cell death. The gp120 causes cross-linking of CD4 molecules without a co-stimulatory signal, resulting in apoptosis.76 Tat upregulates caspase-8 and Fas-ligand in uninfected cells.76,77 Activation- induced cell death (AICD) is also thought to participate in HIV-induced CD4 apoptosis through upregulation of Fas and Fas-ligand.78 Alternatively, AICD may be secondary to the other mechanisms that HIV uses to upregulate Fas and Fas-ligand.79 All of these mechanisms may be further amplified by HIV-mediated cytokine dysregulation.71
HIV-Mediated Cell Killing (Cytopathic Death)
Several theories exist as to how HIV exerts its cytopathic effect on CD4+ cells, leading to direct induction of cell death. Most experts agree that when active HIV replication occurs, the host cell dies. It is still not completely clear how HIV causes this cytopathic effect. Accumulation of HIV DNA in the infected cell is one mechanism postulated to be involved in the direct killing of CD4. During the replication cycle of HIV and other retroviruses, the RNA genome is transcribed into an unintegrated DNA form by a reverse transcriptase. This sequence is usually integrated into host DNA. In animal models of other retroviral infections, unintegrated viral DNA rarely accumulates in infected cells. When DNA accumulates, its presence correlates with the cytopathic effect of the infecting retroviruses. Early evaluations of cells infected by HIV demonstrated that, unlike other retroviruses, accumulation of unintegrated viral DNA seemed to be a more common event. This observation prompted the theory that like other retroviral systems, this DNA accumulation could be responsible for CD4+ cell killing by HIV.80,81 Subsequent studies, however, indicated that accumulation of unintegrated DNA is not directly coupled to cytopathic effect of HIV on CD4+ cells. This was demonstrated by introducing zidovudine or neutralizing antibody against HIV into in vitro systems, where it was observed that CD4+ cell death still occurred despite cessation of accumulation of unintegrated viral DNA genome, effectively demonstrating that unintegrated DNA accumulation was not necessary to induce direct cell death.82,83
Disruption of cellular integrity by HIV has also been postulated to play a role in the direct killing of CD4+ cells. Cellular permeability may be increased and the integrity of the CD4+ cells’ membrane may be compromised during budding of HIV virions from infected CD4+ cells.81 It is also believed that viral products participate in the disruption of the CD4+ membrane. The VPU protein product of HIV causes increased permeability of cells and may also participate in membrane disruption and cell death.84
Interactions between the CD4+ receptor and viral envelope may also participate in the direct cytopathic effect of HIV on T lymphocytes. Two theories exist as to how the envelope protein of HIV interacts with CD4+ receptors to cause cell death. Both CD4+ and the gp160 protein of HIV that codes for the envelope proteins of the virus are processed in the endoplasmic reticulum. This localization of receptor and antigen in the endoplasmic reticulum allows for binding of CD4+ and the envelope protein intracellularly. This receptor-ligand complex may be directly cytopathic.85 Alternatively, the binding of CD4+ in the endoplasmic reticulum decreases the surface expression of CD4+, the receptor for the MHC class II molecule that presents antigen to CD4+ cells to allow activation.86
The HIV envelope protein may directly cause cell death.87 Alterations in the viral envelope were demonstrated to decrease the direct cytopathic effect of the virus without altering the ability of the virus to infect CD4+ cells.88,89 A subsequent study challenged the role of the envelope protein in T cell destruction by engineering a virus with and without the envelope protein. CD4+ cells infected with either of these viruses demonstrated significant cell death independent of the presence or absence of the envelope protein. Though both viruses caused a cytopathic effect, cell death was accelerated more in envelope-positive virus-infected cells than in envelope-negative infected cells.90
Syncytia Formation
One consequence of HIV infection is the formation of CD4+ cell syncytia. Specifically, these syncytia are defined as the fusion of infected and uninfected CD4+ cells to form multinucleated giant cells that ultimately result in cell death and depletion of CD4+ T cells. Early work showed that CD4 molecules were important in syncytia formation by demonstrating decreased formation of these multinucleated cells in the presence of antibody against CD4. Additionally, CD4+ cells do not have to be infected by HIV to be involved in syncytia formation.91
The role of CD4 expression in the formation of syncytia is likely related to the envelope protein (gp120) of HIV. The expression of gp120 on the surface of infected cells may allow for interactions with the CD4 receptor on uninfected cells, leading to formation of syncytia and accelerated CD4+ cell depletion.92 In addition, interaction between cell surface CD4 and HIV gp120 requires other cell adhesion molecules, which may also play a role in the formation of these multinucleated complexes. One such adhesion molecule is leukocyte adhesion receptor-1 (LFA-1). In vitro introduction of an antibody targeting this adhesion molecule inhibits production of HIV-induced multinucleated cells.93 Additional confirmation that LFA-1 is necessary in syncytia formation was supported by identification of a subject with LFA-1-deficient T lymphocytes. In this system, the lack of expression of LFA-1 resulted in the lack of formation of syncytia. Infection of these LFA- 1-deficient CD4+ lymphocytes by HIV, however, was unaffected by the lack of expression of this protein on the surface of these CD4+ T cells.94
The strain of HIV involved may also influence the frequency and occurrence of syncytia. A shift toward the CXCR4 co-receptor-requiring T lymphotropic strains of HIV, also known as syncytium-inducing (SI) virus, in later stages of disease may favor formation of multinucleated cells and enhance depletion of T helper cells. In simian models, shifts from the macrophage-tropic strain of SIV to more T lymphotropic strains of the virus were demonstrated in later stages of disease. This observation raises the possibility that phenotypic shifts in human virus late in infection may accelerate immunodeficiency, perhaps by the participation of the lymphotropic strain in formation of syncytia, depletion of CD4+ cells, and disease progression.95 Clinically, though not an independent predictor of death, the presence of the syncytium-inducing (SI) phenotype of HIV may be a strong predictor of decline in CD4+ cell count and, ultimately, disease progression.96 Despite these observations, syncytia are rarely seen in lymphoid tissue of infected patients, calling to question the in vivo significance of this mechanism of CD4+ cell depletion.96
Antibody-Dependent Cell-Mediated Cytotoxicity
Antibody-dependent cell-mediated cytotoxicity (ADCC) is a process that links the humoral and cellular immune responses. Antibodies directed against the surface of a cell serve as the target of natural killer (NK) cells that carry receptors for the Fc portion of the antibody and are able to then kill the antibody-coated target cell. ADCC may be one of the mechanisms of CD4+ T helper cell depletion in HIV-infected individuals.
In one in vitro study, both infected and uninfected CD4+ cells were coated with gp120 and were exposed to gp120-reactive human sera. The human sera were able to direct an ADCC response against both infected and uninfected CD4+ cells coated with gp120.97 This observation raised the possibility that ADCC may be a pathway that allows for clearance of HIV-infected cells but, by so doing, may also participate in the depletion of HIV-specific CD4+ cells. Excess soluble gp120 produced from infected cells may be able to coat uninfected CD4+ cells, making them targets of in vivo ADCC. An association was also observed between an antibody response against the HIV protein Nef and disease progression. Similar to gp120 discussed above, efficient ADCC was demonstrated against Nef, potentially linking the presence of a Nef antibody response with increased ADCC-mediated CD4+ cell death and HIV disease progession.98
Cytotoxic T Lymphocyte Directed CD4+ Lymphocyte Depletion
HIV-specific cytotoxic T lymphocytes are postulated to play a very important role in controlling HIV during acute infection and during the more latent phases of HIV progression.99 One important function of CD8+ CTLs is the killing of cells infected with HIV. It is, therefore, possible that the CD8+ CTL response thought to be important in controlling infection may lyse HIV-specific CD4+ cells. They may also play a role in dismantling the delicate architecture of the lymphoid organs that allow for an orchestrated immune response to occur against a given antigen.100 There is additional evidence that HIV-infected individuals harbor a population of CD8+ cytotoxic T lymphocytes that participate in depletion of non-HIV-infected “bystander” CD4+ T cells.101
Other Mechanisms of CD4 Depletion and Viral Evasion
Although CD4 depletion likely plays a role in the loss of immune activity against HIV, MHC class II- restricted epitope variation may also play an important role in HIV pathogenesis. In one study, four variant gp120 HIV Th cell epitopes were presented to cells that previously demonstrated a proliferative response against wild-type sequence. Three of the tested variants did not induce proliferation. The fourth variant stimulated attenuated proliferation compared with the wild-type epitope. Pre-exposure of the previously responsive CD4+ T cell clone with variant peptide followed by stimulation with wild-type peptide resulted in inhibition of proliferation as well as cytotoxicity. Inhibition was reversed or prevented by exposing the CD4 clone cells to IL-2.102 The characterization of these epitope variants and their roles in T helper cell immune response is limited. Other models of infection, both human and murine, have demonstrated that sequence variation may alter cellular immune response by decreasing recognition or presentation of MHC class II-restricted epitopes or shifting T helper cell phenotypes from the more protective Th1 response to the less protective Th2 response (Table 12.1).103-105