Interstitial Lung Disease
GENERAL PRINCIPLES
Definition
ILDs are a heterogeneous group of >200 disorders characterized by infiltration of the lung interstitium by cells, fluid, and/or connective tissue.
ILDs can present acutely or chronically, and they are often diagnosed using a multidisciplinary approach employing pulmonary clinicians, radiologists, and pathologists.
Classification
• ILDs can be broadly classified into those with known causes and those without (idiopathic).
î Idiopathic interstitial pneumonias: 59
■ Idiopathic pulmonary fibrosis (IPF) (idiopathic usual interstitial pneumonia [UIP])
■ Idiopathic nonspecific interstitial pneumonia (NSIP)
■ Desquamative interstitial pneumonia (DIP)
■ Respiratory bronchiolitis-associated interstitial lung disease (RB-ILD)
■ Cryptogenic organizing pneumonia (COP) (idiopathic OP)
■ Acute interstitial pneumonia (AIP)
■ Lymphoid interstitial pneumonia (LIP) (rare)
■ Idiopathic pleuroparenchymal fibroelastosis (rare)
î Medication/therapy induced:
■ Bleomycin
■ Amiodarone
■ Nitrofurantoin
■ Checkpoint inhibitors
■ NSAIDs
■ Thalidomide
■ Rituximab
■ Azathioprine
■ Methotrexate
■ Radiationtherapy
î CTD-ILD:
■ Rheumatoid arthritis
■ Scleroderma
■ Sjogrensyndrome
■ Antisynthetase syndrome
■ Mixed CTD
■ Systemic lupus erythematosus
î Vasculitides:
■ Granulomatosis with polyangiitis
■ Eosinophilic granulomatosis with polyangiitis
■ Microscopic polyangiitis
■ Goodpasture syndrome
î Pneumoconiosis (diseases of the lung due to dust inhalation):
■ Coal miners' pneumoconiosis
■ Asbestosis
■ Silicosis
■ Siderosis
■ Stannosis
■ Mixed dust pneumoconiosis
î Granulomatous ILD:
■ Sarcoidosis
■ Berylliosis
■ Hypersensitivity pneumonitis (HP)
■ Granulomatous-lymphocytic interstitial lung disease
■ Bronchocentric granulomatosis
î Cystic lung diseases:
■ Lymphangioleiomyomatosis (LAM)
■ Pulmonary Langerhans cell histiocytosis (PLCH)
■ Birt-Hogg-Dube (BHD) syndrome
■ Pulmonary amyloidosis
■ Light chain deposition disease
■ Postinfectious
î Miscellaneous:
■ Erdheim-Chester disease
■ Pulmonary alveolar proteinosis
■ Lipoid pneumonia
■ Pulmonary alveolar microlithiasis
■ Acute eosinophilic pneumonia
■ Chronic eosinophilic pneumonia
Clinical Presentation
HISTORY
• Obtaining a thorough history is of paramount importance in patients presenting with ILD and is often crucial in making a diagnosis.
• Patients most often present with progressive dyspnea and persistent dry cough.
• Duration of symptoms may help in differentiating ILDs. While many ILDs present with years of progressive dyspnea and cough, a subset of ILD patients present with acute or subacute onset of symptoms (AIP, acute eosinophilic pneumonia, OP), which may mimic infectious pneumonias with atypical organisms.
• Past medical history is very important, not only for underlying diseases but also to identify ILDs related to disease management. Examples include CTDs and immunosuppressive agents; cancers along with chemo-, immuno-, and radiotherapies; and other systemic diseases that can potentially affect the lungs such as inflammatory bowel disease. It is important not to forget the use of over-the-counter medications.
• Documenting a smoking history is essential. Some ILDs manifest almost exclusively in smokers (Langerhans cell histiocytosis, DIP, and RB-ILD). Some diseases are strongly associated with current or previous tobacco use, for example, IPF. Pulmonary hemorrhage is far more common in active smokers with Goodpasture disease than in prior or nonsmokers.
• Exposures both at home and in the workplace should be evaluated. These may include exposures to radiation, asbestos, metal dusts, wood dusts, chemicals or fumes, pets, moldy environments, down comforters and/or pillows, and more. Patients should be questioned regarding the degree and duration of their exposures, and the use of respiratory protective equipment.
• Family history should be obtained, specifically as it relates to pulmonary fibrosis, lung disease, or autoimmune disease. Multiple inheritance patterns have been described with ILDs including complex (sarcoidosis), autosomal dominant (tuberous sclerosis), and autosomal recessive (Hermansky-Pudlak syndrome).
PHYSICAL EXAMINATION
• Extrapulmonary examination in patients with ILD should pay particular attention to findings of systemic diseases that may affect the lungs.
These include CTDs, sarcoidosis, tuberous sclerosis, and others.î Examples include sclerodactyly, mechanic’s hands, Raynaud phenomenon, dry mucous membranes, telangiectasias, skin rashes, facial erythema, papules, eczema, or other skin lesions.
• Cardiac examination should focus on findings suggesting the presence of PH/cor pulmonale including a right ventricular heave, pulmonary artery tap, tricuspid regurgitation holosystolic murmur, right-sided S3, and peripheral edema. These findings are usually indicative of advanced lung disease.
• Clubbing is a very nonspecific finding described in lung diseases, heart diseases, and gastrointestinal (GI) diseases. It can be seen in IPF, sarcoidosis, PLCH, and other ILDs.
• The pulmonary examination in ILDs is nonspecific. Findings may include dry inspiratory crackles, which are best noted posteriorly near the lung bases. Wheezes and inspiratory squeaks may also be noted.
Diagnostic Testing
Diagnostic testing in the evaluation of ILDs typically involves:
• Chest imaging including CXR and high-resolution CT (HRCT) of the chest
• Pulmonary function tests (PFTs)
• Blood testing
• Lung sampling including bronchoscopy and video-assisted thoracic surgery (VATS)
CHEST IMAGING
• Review of old CXRs is often very helpful in assessing both the rate and extent of change of lung disease over time.
• Up to 10% of CXRs may be normal in patients with ILD; as a result, a normal CXR may not exclude ILD in settings where the clinical suspicion of ILD is very high.
• As a result of the wide variety of ILDs, CXRs may have a highly variable appearance.
• The most common abnormality on CXR in ILD is a reticular pattern of linear opacities that may be localized or form a network involving the lungs diffusely.
• However, nodular opacities, alveolar opacities, mixed alveolar opacities, and reticular opacities and occasionally cystic changes are also seen.
• Extensive fibrosis of the lungs may lead to volume loss in one or both lungs.
• Patients with known or suspected ILDs should undergo CT scanning as the diagnosis of many ILDs relies on HRCT. 60
• HRCT is a scanning technique that uses thin slice (usually 1-mm thick) images that are obtained and processed using a high-frequency reconstruction algorithm. 60 Scans are obtained with the patient in a supine position during a breath hold at maximal inspiration and then during a breath hold at maximal expiration.
• Prone imaging may be performed in cases where atelectasis obscures the posterior lung bases.
• The pattern on HRCT is important in determining the differential diagnosis of ILD (Table 10-6). Notably, published guidelines exist for the definitive radiologic diagnosis of certain ILDs, specifically IPF. 60
TABLE 10-6
CLINICAL AND RADIOLOGIC FEATURES OF INTERSTITIAL LUNG DISEASES
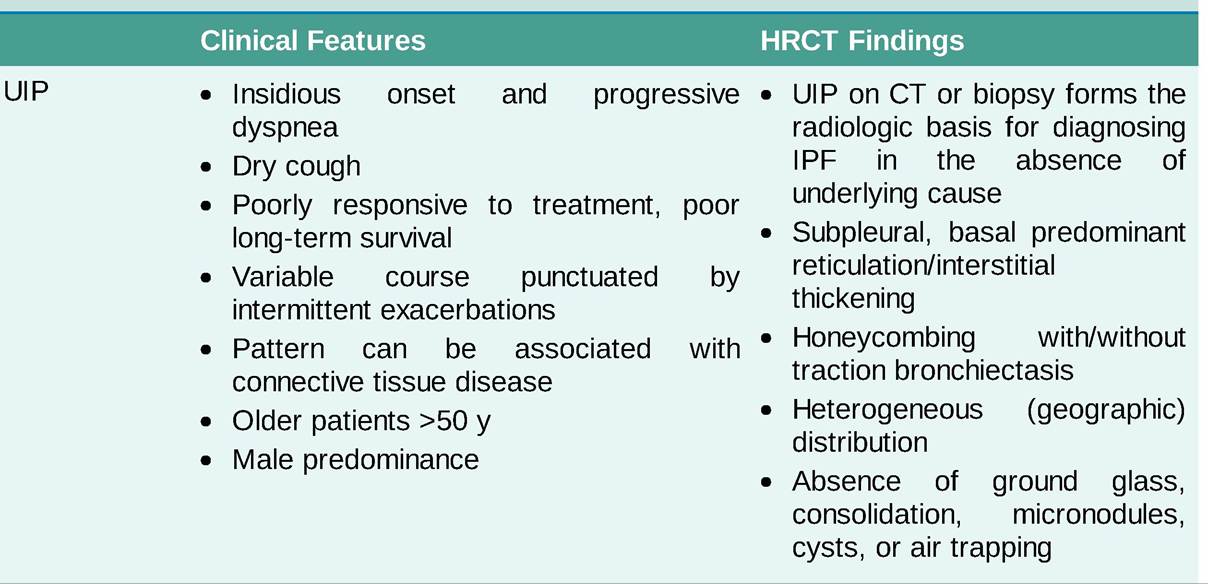


BAL, bronchoalveolar lavage; COP, cryptogenic organizing pneumonia; DIP, desquamative interstitial pneumonia; HP, hypersensitivity pneumonitis; HRCT, high-resolution CT; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; NSIP, nonspecific interstitial pneumonia; RB-ILD, respiratory bronchiolitis-associated interstitial lung disease; UIP, usual interstitial pneumonia.
Data from Kadoch MA, Cham MD, Beasley MB, et al. Idiopathic interstitial pneumonias: a radiology-pathology correlation based on the revised 2013 American Thoracic Society-European Respiratory Society classification system. Curr Probl Diagn Radiol. 2015;44:15-25; Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis - evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med.
2011;183:788-824; Webb RW, Higgins CB. Thoracic Imaging: Pulmonary and Cardiovascular Radiology. Lippincott Williams & Wilkins; 2005.PULMONARY FUNCTION TESTING
• PFTs are a noninvasive set of tests that allow for evaluation of lung function, stratification of disease severity, and for monitoring of disease progression over time.
• Complete pulmonary function testing involves spirometry, lung volumes, and diffusing capacity (DLCO), along with resting and exercise pulse oximetry.
• PFTs allow us to determine whether patients have restrictive lung disease, obstructive lung disease, or a mixed pattern of disease. This aids in the differential diagnosis of ILD.
• While most fibrotic ILDs demonstrate a restrictive pattern on PFTs, a number of ILDs may demonstrate obstruction, including smoking-related lung diseases (RB-ILD and PLCH), cystic lung diseases (LAM and BHD), and sarcoidosis.
• DLCO is commonly decreased in patients with ILD but is a nonspecific finding.
• In some cases, the presence of coexisting COPD in patients with ILDs may lead to a mixed pattern of lung disease on PFTs. In other cases, pseudonormalization of PFTs occurs where the combination of restriction and obstruction leads to the finding of “normal” testing. In these cases, a low DLCO may be the only clue that the patient has significant underlying lung disease when the PFTs are reviewed in isolation.
• Resting and exercise pulse oximetry are commonly assessed using a 6MW test.
• Serial PFTs obtained at follow-up visits are a useful way to monitor for disease progression and may have prognostic significance. 61
LABORATORIES
• Routine laboratory testing in the evaluation of ILD patients includes CBC, BMP, and LFT testing. These may provide clues to the diagnosis (eosinophilia).
• Many drugs used in the treatment of ILD require regular monitoring of blood counts, renal function, or liver function.
• Serologic testing for CTDs is obtained in all patients with clinical stigmata of CTD.
• Aldolase and creatinine kinase may be tested to evaluate for evidence of myositis in patients with a clinical suspicion for antisynthetase syndrome. In cases where suspicion is high for myositis, panels of muscle-specific antibodies should also be obtained.
• In any patient with a high suspicion for scleroderma or Sjogren disease, an ENA panel should be obtained. ENA panels vary but contain a wide variety of antibodies to screen for CTD.
• In practice, serologic testing is often obtained in a wide range of patients to exclude subclinical CTD.
Many centers obtain a minimum of an antinuclear antibody (ANA), rheumatoid factor, and a cyclic Citrullinated peptide antibody tests, even patients with suspected IPF, since rheumatoid arthritis ILD often presents with a UIP pattern on HRCT.
• In patients with cystic lung disease on imaging, testing for vascular endothelial growth factor type D (VEGF-D) may be helpful in making the diagnosis of LAM. Levels of VEGF-D > 800 pg/mL in the correct clinical or radiographic setting can be diagnostic of LAM. 62
• Genetic testing can also be helpful in cystic lung diseases and can screen for tuberous sclerosis- associated LAM (TSC1 and TSC2 gene mutations) and BHD (FLCN, folliculin gene mutations) where appropriate.
LUNG SAMPLING
• When an extensive evaluation does not result in a confident diagnosis, lung sampling can be considered.
• Bronchoalveolar lavage (BAL) is used to sample the cellular content of the lungs. It has limited utility in the evaluation of ILD.
• BAL is useful in excluding coexisting infection and, in some cases, malignancy. It is also useful in cases where diffuse alveolar hemorrhage or eosinophilic lung disease is suspected. 63
• Lung biopsy should only be undertaken at centers with expertise in evaluating ILD patients. Generally, biopsy should be reserved for circumstances where the diagnosis is uncertain and clarification would result in a significantly altered approach to management.
• While lung biopsy is desired in many cases, patients with ILD are often considerably physiologically impaired and may not tolerate the procedure.
• Although many patients tolerate lung biopsy well, certain subgroups of patients are predisposed to complications, including decompensation of their ILD following lung biopsy. 64 Patients with IPF may develop disease exacerbations following lung biopsy, resulting in disease progression and even death.
• Two types of lung biopsy are available: transbronchial forceps biopsy (TBBx) and VATS biopsy.
• TBBx is often performed along with BAL during bronchoscopy. It is most useful in cases in which small biopsy samples suffice for diagnosis.
• TBBx has the highest yield in bronchiolocentric ILDs, such as sarcoidosis, berylliosis, and lymphangitic carcinomatosis. 65 It is also useful in cases where eosinophilic pneumonia or pulmonary alveolar proteinosis is suspected.
• TBBx is insufficient for differentiating most idiopathic ILDs, especially between UIP and NSIP, given inadequate sample size.
• However, a recently developed test, the Envisia Genomic Classifier, uses genomic patterns in TBBx samples to distinguish UIP fibrotic lung disease from non-UIP fibrotic lung disease. It is may helpful in well-selected patients with lung fibrosis who would not tolerate VATS biopsy.
• Transbronchial cryobiopsy is a newer option that allows for larger volume tissue sampling without a surgical lung biopsy. Further studies are required to integrate this technique into the ILD diagnostic algorithm. 66
• In cases with UIP patterns, VATS biopsies are preferred as they yield tissue samples large enough for accurate diagnosis. HRCT should be used to target areas of active disease and avoid lung regions with end-stage fibrosis, which is nondiagnostic.
Specific ILDs
Idiopathic Pulmonary Fibrosis
• IPF is the most common form of idiopathic interstitial pneumonia.
• The incidence of IPF in the US is estimated to be 7-16 cases per 100,000 population. In older populations (e.g., >65 years), the incidence goes up considerably. 67
• The pathophysiology is incompletely understood, but alveolar epithelial cell injury and dysregulated tissue repair are thought to play a significant role.
• A history of cigarette smoking is the strongest risk factor associated with IPF. Other risk factors for IPF include age >60 years, male sex, and GERD. 60
• Increasingly, familial clusters of IPF are being identified and are referred to as familial pulmonary fibrosis (FPF).
• Genetic variant associated with FPF includes those in pulmonary surfactant protein C (SFTPC), surfactant protein A2 (SFTPA2), and mucin 5B (MUC5B). 68
• Around 15% cases of FPF have short telomere syndrome resulting from mutations in telomere maintenance genes including telomerase RNA component (TERC), telomerase reverse transcriptase (TERT), and others. Short telomere syndrome can present with bone marrow failure, premature graying, cirrhosis, nail dystrophy, and mucosal leukoplakia. 69
• Patients with IPF are usually aged 60 years or older.
• They commonly present with slowly progressive dyspnea and nonproductive cough over years to months.
• Extrapulmonary or systemic symptoms are rare.
• Physical examination may reveal dry inspiratory crackles.
Digital clubbing has been reported in 45%-75% of cases depending on the series. 70
• A diagnosis of IPF requires:
î exclusion of all other causes of fibrosing lung disease (CTD, HP, sarcoid) and
î a radiographic pattern of definite UIP on HRCT (Table 10-6, Figure 10-3) or
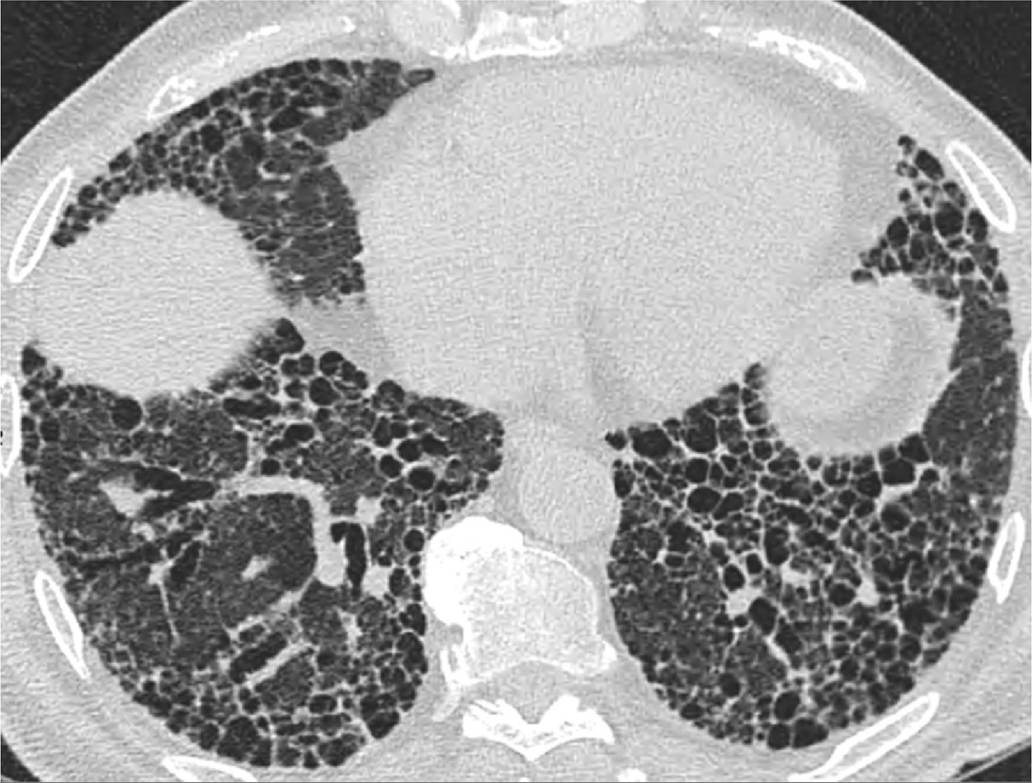
Figure 10-3 Usual interstitial pneumonitis.(Figure courtesy of Dr. Constantine Raptis, Mallinckrodt Institute of
Radiology.)
î a UIP pattern on surgical lung biopsy.
• For patients requiring lung biopsy to confirm the diagnosis of IPF, VATS lung biopsy is preferred, as tissue sampling TBBx is diagnostic in less than one-third of cases. 71
• A multidisciplinary approach employing discussion with pulmonary, radiology, and pathology staff with ILD experience increases diagnostic accuracy.
• Radiographic pattern:
î HRCT is mandatory for imaging ILDs and particularly IPF.
î A pattern of definite UIP on HRCT includes the presence of honeycombing, and subpleural and basilar predominant reticulation. The affected portions of the lung should demonstrate a geographic or heterogeneous involvement. The presence or absence of traction bronchiectasis does not impact the diagnosis.
î The pattern of a definite UIP on HRCT in the appropriate clinical setting (where other potential causes of ILD have been ruled out) may be sufficient to determine the diagnosis of IPF.
î In the setting of a FPF, the HRCT pattern may be atypical, often lacking a basal predominance. Even histologically, strictly defined UIP is identified in less than half of FPF cases. 72
• Disease-modifying treatment options are limited in IPF (Table 10-7).
î There is no medical cure for IPF.
TABLE 10-7
MEDICAL TREATMENT OF SELECTED INTERSTITIAL LUNG DISEASES
| ILD | Potential Therapeutic Interventions a |
| Medication-induced ILD | • Discontinue culprit medication • Corticosteroids |
| Connective tissue disease- | • Corticosteroids |
| associated ILD (UIP, NSIP, COP) | • Immunosuppressive therapy (e.g., cyclophosphamide, azathioprine, mycophenolate, rituximab) |
| IPF | • Pirfenidone • Nintedanib • Consideration for participation in a clinical trial |
| DIP, RB-ILD | • Smoking cessation • Corticosteroids (likely of limited benefit) |
| Sarcoidosis | • Corticosteroids • Immunosuppressive therapy (e.g., methotrexate, azathioprine, infliximab) |
| HP | • Avoid offending antigens • Corticosteroids (likely of limited benefit) • Antifibrotic therapy • Immunosuppressive therapy |
aLung transplantation is a consideration for select patients with end-stage interstitial lung disease.
COP, cryptogenic organizing pneumonia; DIP, desquamative interstitial pneumonia; HP, hypersensitivity pneumonitis; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; NSIP, nonspecific interstitial pneumonia; RB-ILD, respiratory bronchiolitis-associated interstitial lung disease; UIP, usual interstitial pneumonia.
° Most medications studied have not been found to impact disease progression and some have proved dangerous (increased risks of death and hospitalization have been associated with combined use of ^-acetylcysteine, azathioprine, and prednisone). 73
î Pirfenidone, an oral antifibrotic agent, and nintedanib, an oral tyrosine kinase inhibitor, have both been shown to slow the rate of lung function decline in patients with IPF. 74, 75 They may also have a mortality benefit.
î The most frequent side effects with nintedanib are diarrhea (62% of patients), nausea, and vomiting.
° The most frequent side effects of pirfenidone are skin rash (30%), photosensitivity, nausea, and diarrhea.
î Drug-induced liver disease can occur with both agents, and monitoring of liver function tests (LFT) is mandatory with both agents. Elevations in LFTs can be managed with dose adjustments or discontinuation.
• Pulmonary rehabilitation has been associated with improvements in 6MW distance and quality of life in IPF. 73
• Prognosis and clinical course are variable, but those diagnosed with mild, moderate, and severe disease by spirometry have been reported to have median survivals of 55.6, 38.7, and 27.4 months, respectively. 74
• Poor prognostic factors for IPF:
î Decline in forced vital capacity of >10% over six months
î Decrease in DLCO of >15% over six months
î Decrease in 6MW distance of >150 m over 12 months 61
• Exacerbations of IPF are characterized by acute worsening of dyspnea or oxygenation (within 30 days) and new ground-glass opacifications/ consolidations on CT with no evidence of infection, pulmonary embolus, or heart failure. 60
• Exacerbations are typically treated with high-dose corticosteroids, although their benefit has not been systematically proven. Patients often do not return to their pre-exacerbation baseline after IPF exacerbations and mortality rates are high. 76
• Lung transplantation remains the ultimate therapy in patients with advanced IPF. Without lung transplantation, outcomes in IPF remain poor. Patients should be referred to a lung transplant program at the time of diagnosis of IPF.
Nonspecific Interstitial Pneumonia
• NSIP can describe both a radiographic pattern on HRCT and pathologic pattern on lung biopsy.
• It is referred to as nonspecific because biopsies, in particular, lack the histologic features characteristic of other idiopathic interstitial pneumonias.
• NSIP is one of the subtypes of idiopathic interstitial pneumonia. However, idiopathic NSIP is rare.
• NSIP is much more commonly secondary to other causes including 77 :
î CTD: Including scleroderma, Sjogren disease, antisynthetase syndrome, and rheumatoid arthritis. Interstitial pneumonia with autoimmune features is a distinct subcategory of ILD where patients with confirmed NSIP on HRCT or lung biopsy have clinical features of autoimmune disease not conforming to any particular CTD. 78
î Drug toxicity: Drugs commonly associated with NSIP including amiodarone, nitrofurantoin, methotrexate, statins, and various chemotherapeutic agents have been associated with development of NSIP.
î HIV infection: Less common in the age of highly active antiretroviral therapy (HAART).
î Others: These include fibrotic HP, FPF, graft versus host disease, and IgG4 disease.
• Patients with NSIP tend to be younger than those with IPF and are more commonly female. 79
• Due to the frequency of secondary causes, NSIP may present with fevers, chills, weight loss, or flu-like symptoms. Symptoms of CTD are also common.
• HRCT demonstrates a combination of ground-glass and reticular opacities often in a peripheral and basal predominant distribution. A peribronchovascular distribution may also be noted. There is often sparing of the immediate subpleural space from involvement, which is relatively specific for NSIP (Table 10-6) (Figure 10-4).
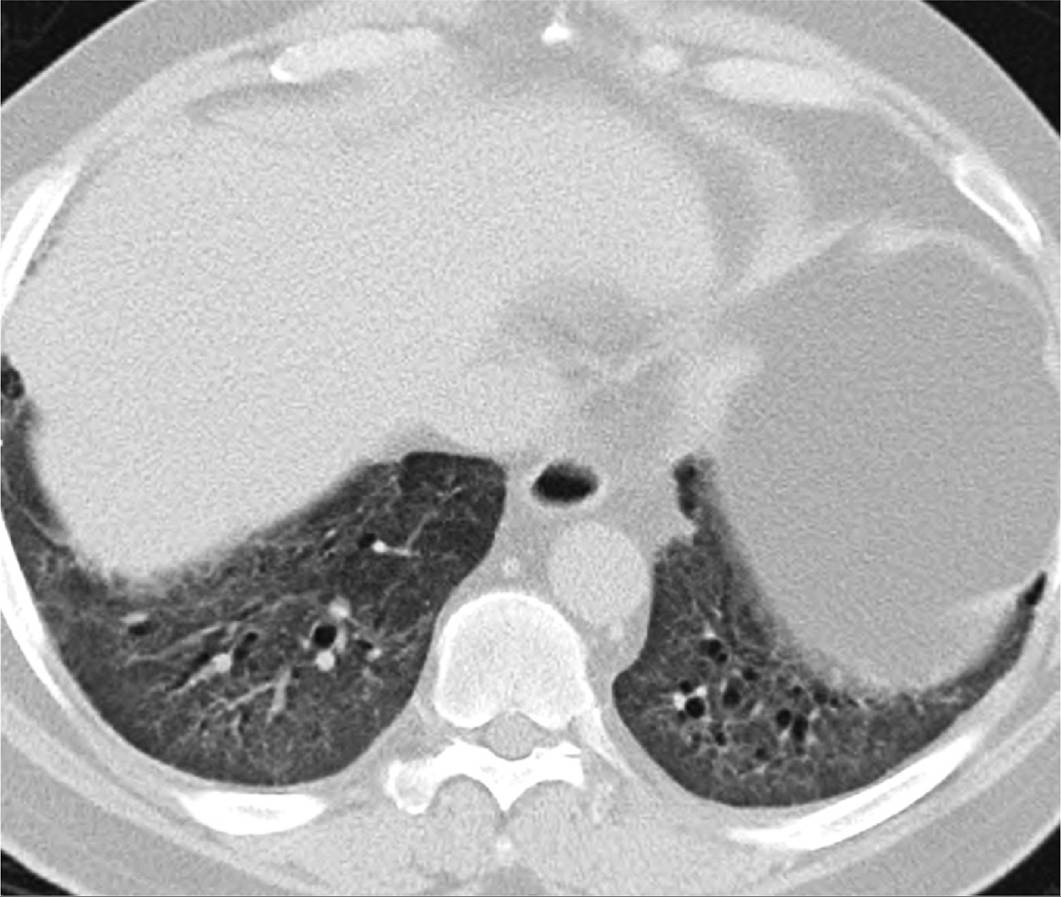
Figure 10-4 Nonspecific interstitial pneumonitis.(Figure courtesy of Dr. Constantine Raptis, Mallinckrodt Institute of
Radiology.)
• Other features of NSIP include traction bronchiectasis, centrilobular nodules, air trapping on exhalation imaging and rarely microscopic honeycombing.
• While the diagnosis of idiopathic NSIP may require a surgical lung biopsy, most cases of secondary NSIP do not when the diagnosis is certain from the history, physical examination, and laboratory testing. In cases where lung biopsy is required, VATS biopsy is essential.
• Treatment of CTD-associated NSIP usually involves immunosuppression with steroids, in combination with steroid-sparing agents such as mycophenolate, azathioprine, rituximab, or cyclophosphamide (Table 10-7).
• Management of patients with CTD-related ILD should be undertaken in a multidisciplinary fashion.
• NSIP from drug toxicity is treated by discontinuation of the offending agent. In some cases, steroid therapy may be required.
• Other secondary causes of NSIP are treated by targeting the primary disease, for example, HIV (HAART) and IgG4 disease (steroids).
• In cases refractory to therapy, lung transplant remains an option in selected instances.
• Patients with NSIP have a significantly better prognosis than those with IPF. 79
Hypersensitivity Pneumonitis
• HP is a pulmonary syndrome of varying clinical presentation and natural history.
• It is the result of an immune-mediated pulmonary response directed against a plethora of potential inhaled antigens to which an individual is both sensitized and hyperresponsive.
• Hundreds of antigens have been described as causing HP and include bacteria, mycobacteria, fungi, organic proteins, chemicals, and metals. 80
• Well-described clinical presentations of HP include farmer's lung (exposure to moldy hay) and bird fanciers' disease (exposure to birds).
• Since a minority of people exposed to inhaled antigens manifest with disease, disease expression is thought to be dependent on a complex interaction of antigen dose, intensity and duration of antigen exposure, antigen immunogenicity, and host factors such as genetic susceptibility.
• Currently, the optimal characterization of HP is based on the presence or absence of fibrosis and honeycombing on either HRCT or pathologic samples. The radiologic phenotypes in particular have prognostic significance 81 :
î HP without fibrosis (median survival >14 years)
î HP with fibrosis (median survival 7.95 years)
î HP with fibrosis and honeycombing (median survival 2.8 years)
• Acute forms of HP (corresponding to nonfibrotic disease) may manifest over hours to weeks and are often temporally related to antigen exposure. They present with relatively rapid onset of dyspnea, cough, and chest tightness. In addition, patients may manifest with systemic symptoms including fevers, chills, myalgias, and malaise. Resolution is expected following antigen removal.
• Chronic disease (corresponding to fibrosis and honeycombing) and dyspnea often progress indolently over time and are often associated with dry cough. Unlike IPF, chronic forms of HP can be accompanied by systemic symptoms including anorexia, weight loss, and fatigue.
• A careful and thorough exposure history should be taken and should include inquiry about exposure to birds or bird feathers (including down pillows and comforters), hot tubs (associated with aerosolized mycobacterial exposure), air humidifiers, moldy homes or workplaces, animal furs, epoxies, plant matter, industrial dusts, and chemicals. 82
• Specific antibody testing for culprit antigens can be sent to specialized labs when indicated, although positive serologies only support exposure to the antigens against which the antibodies are directed and do not necessarily confirm causation.
• The causative exposure/antigen is identified in 20% of cases.
î Endocrine involvement can manifest as hypercalcemia and hypercalciuria secondary to dysregulated production of calcitriol. 90
• Sarcoidosis may also present with two well-described acute clinical syndromes 88 :
î Lofgren syndrome is characterized by arthritis, erythema nodosum, and bilateral hilar lymphadenopathy. 86
î Heerfordt syndrome presents with a combination of uveitis, parotid gland swelling, fevers, and in some cases facial palsy. It is also known as uveoparotid fever.
• CXR imaging in sarcoidosis may manifest with pulmonary opacities, thoracic lymphadenopathy, or a combination of both. CXR is also used to stage the disease (Table 10-8).
TABLE 10-8
Scadding staging of sarcoidosis


Adapted from Maller V, Knipe H. Thoracic sarcoidosis (staging). Accessed March 4, 2021. http://radiopaedia.org/articles/thoracic- sarcoidosis-staging?lang=us
• On HRCT, parenchymal nodules appear in almost 80% of patients. They typically follow a perilymphatic distribution and may coalescence into larger opacities. 86 Other findings may include alveolar opacities, pulmonary fibrosis, and air trapping on expiratory imaging (Table 10-6).
• Laboratory testing should be obtained. Serum calcium, urine calcium, CBC, BMP, and LFTs should all be checked. Abnormalities in these are not diagnostic of sarcoidosis but can help determine extent of disease and organ involvement.
• In the appropriate clinical setting, sarcoidosis is diagnosed by the presence of noncaseating granulomas on biopsy samples of involved organs (commonly the lung, lymph nodes, or skin). The presence of granulomas in more than one organ system is preferable when making the diagnosis. 90
• Exclusion of other diseases is obligatory when diagnosing sarcoidosis. 90 Excluding infectious disease is of particular importance, especially in patients who reside in areas with endemic fungal or mycobacterial disease, as these can mimic sarcoidosis.
• Treatment of sarcoidosis can be complicated, and referral to pulmonary and other specialists is often necessary.
• For mild disease, symptoms and radiographic changes may remit in the absence of treatment.
• In the setting of more symptomatic or progressive disease, corticosteroids are typically first-line therapy and many patients can be treated with intermittent steroid therapy alone (Table 10-7).
• Patients with more advanced disease or those requiring longer-term steroid therapy may need to be transitioned to steroid-sparing immunosuppression such as methotrexate or azathioprine.
• Tumor necrosis factor alpha antagonists are typically reserved for severe disease that progresses despite the aforementioned therapies. 91
• Prognosis is highly variable, ranging from indolent self-remitting disease to progressive fibrosis requiring transplantation. As a broad rule, acute onset disease tends toward a better prognosis, while more indolent onset disease may become unremittingly progressive.
Organizing Pneumonia
• OP is a nonspecific pulmonary response to injury characterized by inflammation and proliferation of granulation tissue in the alveoli and terminal bronchioles.
• OP was previously referred to as bronchiolitis obliterans with OP.
• OP may be either:
î Idiopathic, referred to as COP, and one of the subtypes of idiopathic interstitial pneumonia, or
î Secondary, and occur in association with CTDs, drug toxicity (e.g., amiodarone, checkpoint inhibitors), infections, inhalational injury (e.g., cocaine, industrial gasses), radiation treatment, and along with other ILDs (e.g., vasculitis). 92
• Patients often present with dyspnea, cough, fevers, malaise, fatigue, and weight loss lasting weeks to months. These symptoms may mimic pneumonia, and a frequent scenario is a patient presenting with multiple episodes of “pneumonia” unresponsive to antibiotics therapy.
• On HRCT, OP manifests with multifocal patchy consolidation, often in a peripheral or peribronchovascular distribution. The opacities may affect all lung zones and when followed over time may be migratory. 92 The reverse halo or atoll sign is thought to be very specific for OP on HRCT but is not commonly seen (Table 10-6).
• Diagnosis is often suggested through a combination of good history taking and HRCT appearance.
• PFTs are nonspecific and may demonstrate restriction.
• BAL is useful to rule out infection.
• The method of lung biopsy is controversial. While TBBx may make the diagnosis, some feel that they may not be sufficient to detect secondary causes of OP due to the small size of the biopsies. As such, several centers recommend VATS biopsy where OP is suspected. 64
• Patients normally have good response to treatment with steroids. However, recurrence is common and long-term steroid therapy over 3-6 months is generally recommended. If OP continues without response to treatment or progresses to fibrosis, prognosis is poor.
• In cases associated with other diseases (e.g., vasculitis, CTD), treatment is directed at the underlying cause.
Smoking-Related ILD
Certain ILDs manifest almost exclusively in smokers. These include RB-ILD, DIP, and PLCH.
RESPIRATORY BRONCHIOLITIS INTERSTITIAL LUNG DISEASE
• RB-ILD is typically associated with heavy smoking (often 30 pack-years or more). 93
• It commonly presents in the third to fifth decades of life.
• On HRCT, RB-ILD manifests with centrilobular ground-glass nodules, often with an upper lobe predilection. In some cases, the nodules are more confluent and present as ground-glass opacities (Table 10-6).
• Other changes related to smoking, such as emphysema and bronchial wall thickening, may also be present. 94
• Symptoms and radiographic findings often improve with smoking cessation (Table 10-7). 94
DESQUAMATIVE INTERSTITIAL PNEUMONIA
• DIP may represent a spectrum of disease along with RB-ILD.
• Ninety percent of patients with DIP are heavy smokers. 95
• The other 10% may have CTD, HIV or environmental exposures. A congenital form may be seen in children.
• The predominant HRCT finding in DIP is bilateral ground-glass opacity, which may be peripheral, patchy, or diffuse in distribution. It is classically described as triangular-shaped opacities radiating from the hila to the periphery of the lung; however, this finding is noted in only a minority of patients (Table 10-6).
• Small cystic spaces occasionally develop within the ground-glass opacities.
• Response to smoking cessation is favorable, although some patients may require corticosteroid therapy (Table 10-7). 96
• The disease occasionally persists despite therapy.
PULMONARY LANGERHANS CELL HISTIOCYTOSIS
• Langerhans cell histiocytosis is a rare disorder of unknown etiology.
• It results from abnormal clonal proliferation of Langerhans cells derived from bone marrow precursors.
• Multisystem involvement with extrapulmonary disease involving the skin, central nervous system, skeleton, and other organs is common in children but rare in adults where disease is confined to the
lungs.97
• PLCH presents in adult smokers between the ages of 20 and 40 years with cough, dyspnea, weight loss, and occasionally spontaneous pneumothorax. 97
• HRCT demonstrates a combination of nodules and cysts. 63
• Nodules tend to predominate in early disease. They range from few to innumerable in number and typically have a centrilobular distribution. They may have irregular margins and cavitate to form cysts.
• Cystic lung lesions predominate in later disease. The cysts are characterized by irregular/bizarre margins and have an upper lobe predominance, often sparing the lung bases. They cysts are commonly thin walled but may occasionally be a few millimeters thick.
• Unlike typical ILDs, lung volumes are often preserved.
• The primary therapy for PLCH is smoking cessation. Response to smoking cessation is considered good, with up to 50% of patients demonstrating improvement or resolution. 98
• Approximately 20% of patients have persistent, progressive disease.
• In these patients, a trail of steroid therapy may be considered, but data for this are lacking.
• In the most resistant forms of disease, chemotherapeutic agents including cladribine and vinblastine have been used with variable success.
• Some patients with PLCH have mutations in the mitogen-activated protein kinase pathway such as BRAF V600E. In these patients, targeted therapy with vemurafenib (a BRAF kinase inhibitor) has been used. 98
Pneumoconioses
• Pneumoconioses are diseases of the lung parenchyma that result from exposure to airborne dusts or fibers including asbestos, silica, beryllium, coal, tin, and others. 99
• Pneumoconiosis (with the exception of asbestosis) are characterized by upper lobe-predominant nodular patterns on CT that have the potential to conglomerate over time to form large space occupying lesions known as progressive massive fibrosis.
ASBESTOS-INDUCED LUNG DISEASE
• Arises from exposure to asbestos, a substance historically used in construction, insulation, and fireproofing materials.
• HRCT findings may range from pleural thickening, pleural plaques (often with calcification), subpleural banding (reticulation running parallel to the pleura), to parenchymal changes resembling UIP. The presence of pleural plaques aids in differentiation from other ILDs, but asbestos-related fibrotic disease can exist in the absence of pleural manifestations.
• Treatment focuses on asbestos avoidance and supportive care.
• Prognosis is good in mild disease, although the risk of lung cancer is significantly increased in the setting of concomitant cigarette use.
• Exposure to asbestos also increases the risk of developing mesothelioma.
SILICOSIS
• Silicosis results from exposure to crystalline silica, which is found in stone and sand. Foundry workers, construction workers, sandblasters, and glassblowers are at increased risk. 100
• HRCT typically demonstrates small nodules in the upper and mid zones with hilar adenopathy in a pattern that may resemble those seen in sarcoidosis. This is known as simple silicosis.
• Simple silicosis may progress to complicated silicosis characterized by coalescence of nodules in the perihilar areas of the lung to form conglomerate masses or areas of progressive massive fibrosis.
• Treatment is supportive, although close monitoring for development of TB is warranted given the increased risk of TB in these patients. 101
• An acute form of silicosis, also known as silicoproteinosis, has been described with episodes of inhalation of high concentrations of silica. It appears as diffuse ground-glass opacities on CXR. Mortality is high.
BERYLLIOSIS
• Berylliosis is caused by exposure to beryllium and beryllium compounds. 102
• Exposure occurs in the aerospace industry, atomic industry, beryllium mining, and fluorescent light bulb manufacturing.
• It is clinically indistinguishable from pulmonary sarcoidosis.
COAL WORKERS’ PNEUMOCONIOSIS
• Coals workers' pneumoconiosis (CWP) is caused by inhalation of high carbon coal dust. It is commonly known as “black lung disease.”
• HRCT typically demonstrates small nodules in the upper and mid zones. This is known as simple silicosis. 103
• Simple CWP may progress to complicated CWP characterized by coalescence of nodules in the perihilar areas of the lung to form areas of progressive massive fibrosis.
• Caplan syndrome, also known as rheumatoid pneumoconiosis, is the presence of pulmonary nodules in the lungs of patients diagnosed with rheumatoid arthritis who have also been exposed to coal dust. It has also been described in patients with rheumatoid arthritis exposed to silica.
Cystic Lung Diseases
• Cystic lung diseases are a heterogeneous group of disorders that include LAM, PLCH, BHD, LIP, pulmonary amyloidosis, and light chain deposition disease.
• These diseases are characterized by the presence of cysts on HRCT imaging. Cysts are air-filled lucencies or low attenuation areas with thin (usually ≤2 mm) walls located within normal lung parenchyma. 104
• The cysts range from few to innumerable in number.
• Cysts should not be confused with emphysema, bullae, pneumatoceles, honeycombing, or cavitary lung lesions.
• The location and thickness of the cyst walls can be helpful for disease differentiation.
î PLCH is generally characterized by upper lobe-predominant cysts with thicker and irregular/bizarre-shaped walls.
î BHD demonstrates larger-sized cysts with a peripheral and basilar distribution, often abutting the pleura.
î LAM, LIP, and amyloidosis most often have a random distribution of cysts. 105
• Cystic lung diseases are associated with a high incidence of pneumothorax compared to the general population, and pneumothorax is a common presenting complaint.
Lymphangioleiomyomatosis
• LAM is a progressive cystic lung disease seen almost exclusively in women of childbearing age.
• LAM may develop sporadically or as part of tuberous sclerosis complex (TSC), a neurocutaneous multisystem disorder characterized by multiple benign hamartomas of the skin, brain, kidney, lung, and other organs. 106
• Men with TSC may also develop LAM.
• Most patients present with progressive dyspnea. Other presentations include pneumothorax, and chylous pleural and abdominal effusions.
• The most common extrapulmonary manifestations of LAM are renal angiomyolipomas (AML)—benign kidney tumors containing blood vessels, smooth muscle, and fatty tissue. 107
• Lymphangioleiomyomas are another characteristic feature of LAM. They are fluid-filled structures that can be seen in the retroperitoneal space, pelvis, and mediastinum. 106
• LAM is usually diagnosed based on a combination of clinical presentation and imaging findings including cystic lung disease and renal AMLs.
• VEGF-D levels can be tested, with levels ≥800 pg/mL reliably distinguishing LAM from other cystic lung diseases. 62
• Genetic testing for tuberous sclerosis (TSC1 and TSC2 gene mutations) can be undertaken where clinically indicated.
• In rare cases, lung biopsy may be needed for histopathologic confirmation. 108 This is usually pursued via bronchoscopy and TBBx. Surgical biopsy is no longer commonly performed. Lesions will demonstrate characteristic human melanoma black 45 staining.
• First-line therapy for pulmonary LAM currently is the mammalian target of rapamycin inhibitor, sirolimus, which has been shown to reduce disease progression. 109
• Everolimus has been shown to be effective in patients who do not tolerate sirolimus. 110
• Otherwise, supportive care, including oxygen therapy, avoidance of activities that could place patients at higher risk for pneumothorax, and pulmonary rehabilitation are mainstays of therapy.
• In some cases, patients may require evaluation for lung transplantation.
OTHER CYSTIC LUNG DISEASES
• PLCH:
î PLCH is described in the “Smoking-related ILD” section.
• BHD syndrome is a rare cause of cystic disease associated with skin and renal neoplasms. 111
î It is the result of germline mutations in the FLCN gene, whose product is folliculin, a putative tumor suppressor protein.
î Pulmonary cysts in BHD are irregularly shaped and commonly localized to the lung bases, often in a subpleural location. 112
î BHD can be diagnosed based on clinical presentation and may be confirmed by skin biopsy showing fibrofolliculomas, a skin hamartoma characteristic of BHD.
î Genetic testing for FLCN mutation can also be performed.
î Treatment is supportive, and progression is generally slow. 107
• Amyloidosis can be associated with cystic lung disease in the setting of underlying systemic amyloidosis or may be organ limited to the lungs (MALT lymphoma). It may also be seen in longstanding CTDs or myeloma. 113
î The cysts are variable in size and distribution. They may be associated with tracheal disease or pulmonary nodules, which often calcify.
î Treatment focuses on the underlying condition.
• LIP is a rare disease, usually associated with CTDs (primarily Sjogren syndrome), lymphoproliferative diseases (e.g., lymphoma), and viral infections (e.g., HIV). Idiopathic cases also occur. 114
î Imaging demonstrates irregular cysts, multifocal ground-glass opacities, nodularity, and septal
thickening.
î Treatment and prognosis are variable depending on the underlying condition.
General Management Considerations
• All ILD patients should be monitored for the development of hypoxemic respiratory failure. Supplemental oxygenation should be provided to maintain oxyhemoglobin saturation by pulse oximetry (SpO2) ≥ 89% both at rest and with exertion (measured by 6MW testing).
• Smoking cessation/avoidance should be strongly encouraged.
• Patients should avoid occupational/environmental triggers of their ILD, if identified.
• Pulmonary rehabilitation therapy should be prescribed for all patients if they meet eligibility criteria.
• Bone density assessment is recommended for patients receiving chronic systemic corticosteroid therapy, along with periodic reassessment (e.g., every 1-2 years).
• Pneumocystis jirovecii pneumonia prophylaxis should be considered in patients receiving chronic steroid therapy, 115 generally at doses of >15 mg prednisone daily.
• Patients on immunosuppressive therapy should have appropriate bloodwork (CBC and/or BMP and/or LFT) monitored periodically.
• Patients should receive vaccinations against pneumococcus and influenza.
• Patients with ILDs should be considered for referral to centers with expertise in the diagnosis and treatment of these conditions, with consideration for participation in ongoing clinical trials.
• ILD increases the risk of PH. 116 Patients with dyspnea out of proportion to their parenchymal lung disease or those with symptoms of right heart failure should be screened with TTE. Although the use of pulmonary vasodilators in this population remains controversial, recent studies have shown improvement in functional status with inhaled treprostinil. 117
• Several ILDs are associated with an increased incidence of malignancy. (e.g., IPF, asbestosis). Rapid weight loss or radiographic changes (e.g., new solitary nodules or persistent consolidation) should raise suspicion and prompt further workup.
• Goals of care and expectations of therapy should be made clear to all patients.
• Palliative care is an ongoing part of disease management in many ILDs, and hospice care should be discussed with all patients with advanced disease who are not transplant candidates. Open discussions of goals of care are helpful in guiding management of acute exacerbations and progressive disease. 118