Thalassemia
GENERAL PRINCIPLES
Definition
The thalassemia syndromes are inherited disorders characterized by reduced Hgb synthesis associated with mutations in either the #945;- or #946;-gene of the molecule (Table 21-3).
TABLE 21-3
THALASSEMIAS
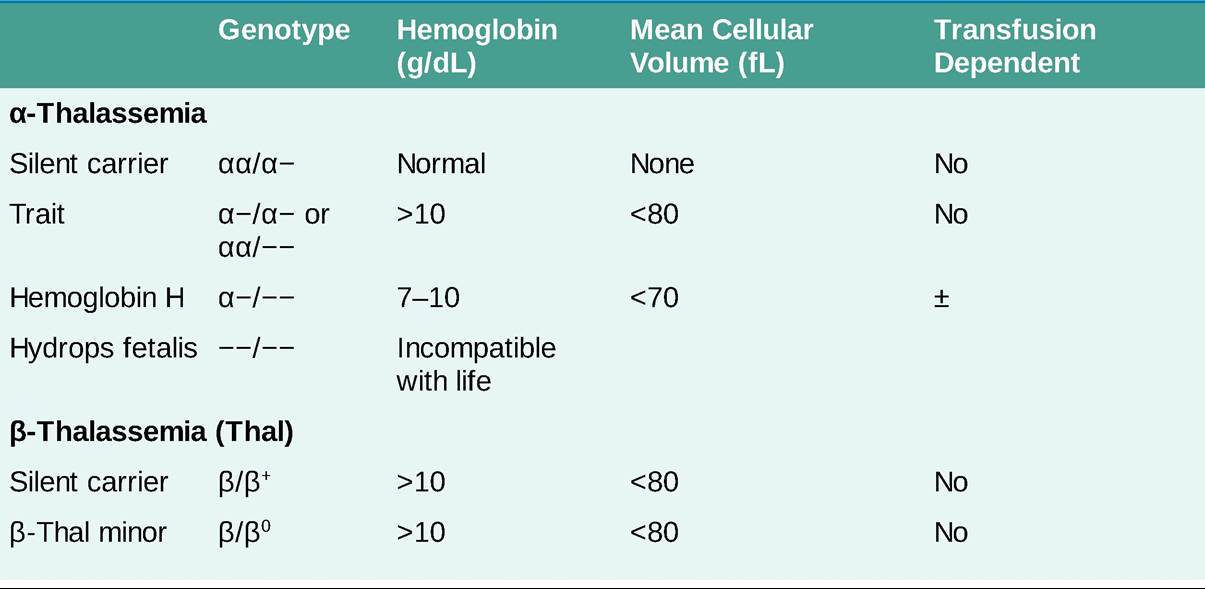

#946;+, #946;-thalassemia genes produce some #946;-globin chains but with impaired synthesis; #946;0, #946;-thalassemia genes produce no #946;- globin chains.
Etiology
• #946;-Thalassemia results in a decreased production of #946;-globin and a resultant excess of #945;-globin, forming insoluble #945;-tetramers and leading to ineffective erythropoiesis.
î #946;-Thalassemia minor (trait) occurs with one gene abnormality with underproduction of #946;-chain globin. Patients are asymptomatic and present with microcytic, hypochromic RBCs and Hgb levels gt;10 g/dL.
î #946;-Thalassemia intermedia (non-transfusion-dependent) occurs with dysfunction in both #946;-globin genes so that anemia is more severe (Hgb 7-10 g/dL).
î #946;-Thalassemia major (Cooley anemia or transfusion-dependent) is caused by mutations of both #946; globin genes that fail to produce significant amounts of #946;-globin and generally require lifelong RBC transfusion support.
• #945;-Thalassemia occurs with a deletion of one or more of the four #945;-globin genes, leading to a #946;-globin excess.
î Mild microcytosis and mild hypochromic anemia (Hgb gt;10 g/dL) are seen with the loss of one or two #945;-globin genes (silent carrier and #945;-thal trait).
î Deletion of three #945;-globin genes (Hgb H disease) results in splenomegaly and hemolytic anemia. In patients with Hgb H disease, transfusion or splenectomy is often not necessary until after the second or third decade of life.
In addition, oxidant drugs similar to those that exacerbate glucose-6- phosphate dehydrogenase (G6PD) deficiency should be avoided because increased hemolysis may occur.î Hydrops fetalis occurs with the loss of all four #945;-globin genes and is incompatible with life.
DIAGNOSIS
• Peripheral smear may show microcytic hypochromic RBCs, along with poikilocytosis and nucleated RBCs.
• Hgb electrophoresis is often diagnostic for #946;-thalassemia showing an increased percentage of Hgb A2 and Hgb F.
• Silent carriers with a single #945;-chain loss generally have a normal electrophoresis. Adults with Hgb H disease demonstrate Hgb H (#946;-tetramers) on electrophoresis. The diagnosis of #945;-thalassemia is confirmed by #945;-globin gene analysis.
TREATMENT
• Patients with either #945;- or #946;-thalassemia trait require no specific treatment.
• In patients with more severe forms of the disease, chronic RBC transfusions to maintain an Hgb level of 9-10 g/dL are needed to prevent the skeletal deformities that result from accelerated erythropoiesis.
• In severe forms of thalassemia, repeated transfusions result in tissue iron overload, which may cause congestive heart failure (CHF), hepatic dysfunction, glucose intolerance, and secondary hypogonadism. Iron chelation therapy delays or prevents these complications. Once clinical organ deterioration has begun, it may not be reversible.
• Chelation therapy is indicated for transfusion-associated iron overload from any cause. It is indicated in patients with a ferritin consistently gt;1000 ng/mL, which may occur after a transfusion burden of gt;20 units of packed RBCs (pRBCs).2
î Deferoxamine, 40 mg/kg SC or IV over 8-12 hours of continuous infusion.
î Deferasirox dispersible tablet 20-40 mg/kg/d (Exjade) is an effective oral chelating agent, but GI disturbances often limit adherence to therapy. Deferasirox film-coated tablet (Jadenu) is available and dosed at 70% of Exjade (7-21 mg/kg/d).
This formulation is often better tolerated. Dose can be titrated every 3-6 months based on ferritin level. Efficacy is similar to that of deferoxamine.• Chelation therapy should be continued until ferritin levels of lt;1000 ng/mL are achieved and maintenance therapy is often needed when RBC transfusions are ongoing. Luspatercept (1 mg/kg every 3 weeks) is approved for transfusion-dependent #946;-thalassemia to reduce transfusion requirements with median time to response 12-24 days.3
• Stem cell transplantation (SCT) is the only curative therapy and should be considered in young patients with thalassemia major who have HLA-identical donors. Gene therapy is the subject of ongoing research and holds promise.4
• Splenectomy should be considered in patients with accelerated (more than two units/month) transfusion requirements. To decrease the risk of postsplenectomy sepsis, immunization against pneumococcus, Haemophilus influenzae, and Neisseria meningitidis should be administered at least 2 weeks before surgery if not previously vaccinated (see Appendix A, Immunizations and Postexposure Therapies). Splenectomy is rarely recommended in patients who are younger than 5-6 years because of the increased risk of sepsis.