Chapter 7 Care of the fetus
Biophysical profile
Cardiotocography
Doppler ultrasound
Fetal abnormalities: cardiovascular
Fetal abnormalities: central nervous system
Fetal abnormalities: chromosomal anomalies
Fetal abnormalities: genetic disorders
Fetal abnormalities: face
Fetal abnormalities: gastrointestinal system
Fetal abnormalities: limbs
Fetal abnormalities: head and neck
Fetal abnormalities: skeletal abnormalities/dysplasias
Fetal abnormalities: thorax
Fetal abnormalities: urinary system
Fetal movement charts
Fetal nuchal translucency
Fetal abnormalities: hydrops
Invasive procedures
IUGR (intrauterine growth restriction)
Multiple pregnancy
Oligohydramnios
Placental abnormalities
Polyhydramnios
Red blood cell isoimmunization
Screening for fetal aneuploidy
Symphyseal fundal height Biophysical profile Definition
The fetal biophysical profile (BPP) score is a real-time ultrasound-based surveillance method that is used to estimate the probability of fetal hypoxaemia at the time of testing.
The five variables that constitute the BPP are affected by the amount of oxygen delivery to the brain and kidney. The advantage of the BPP is its ability to evaluate for both acute and chronic fetal compromise (Manning 1984).This score consists of five parameters: fetal tone, gross fetal movements, fetal breathing movements, amniotic fluid volume (principally related to fetal urine output), and fetal heart rate reactivity (Table 7.1.1). Pathophysiology
The fetal dynamic variables examined in the BPP are dependent on the activity of their regulatory centres in the brain, the integrity of efferent nervous connections, and an intact effector peripheral apparatus. The activity of these connections can be modulated by physiological mechanisms and fetal disease (Manning 2002).
The most important physiological modifier of fetal activity is physiological variation of its behavioural state.
During active states dynamic variables can be observed in a short observation period, while in resting states there may be long periods of inactivity. This is particularly common beyond 34 weeks as fetal rest periods become more frequent.Pathological conditions that can modulate dynamic variables include chronic hypoxaemia, acidaemia, anatomic defects leading to disruption of neural pathways, and medications that interfere with neurotransmission.
Because individual biophysical variables may be absent due to physiological as well as pathological conditions, five variables have been combined to provide the most accurate prediction of fetal health. To account for behavioural states the BPP is scored over a 30-minute interval and may even be extended over 1 hour near term.
Fetal heart rate variables provide a record of autonomic regulation of intrinsic cardiac activity and its modulation by regulatory centres. The main regulatory centres are the vasomotor centre, reticular activating system, and autonomic nervous system. For the BPP the fetal heart rate reactivity is assessed visually by gestational age-graded criteria.
Table 7.1.1 Biophysical profile score (30 minutes)

Amniotic fluid production is dependent on fetal urination and therefore renal plasma flow as well as fetal fluid balance. Since these parameters are dependent on oxygenation, placental fluid exchange and fetal cardiovascular status, the amniotic fluid volume is the only parameter in the BPP that allows longitudinal assessment for chronic fetal deterioration. Clinical management protocol
The clinical use of the BPP also predicates the appropriate management steps for each score. These recommendations are based on the perinatal morbidity and mortality associated with each score and the accuracy of predicting prelabour acidaemia. The five-component BPP provides an accurate assessment of fetal acid–base status from 20 weeks onward.
One parameter that requires special mention is fetal breathing, which is principally determined by maternal/fetal glucose levels: the absence of fetal breathing should be re-evaluated after correction of maternal fasting.Normal BPP
• Score 10/10, 8/10 (normal AFV), and 8/8 (non stress test not done); normal test result. Fetus is not compromised at the time of testing. The risk for unexplained stillbirth in the week following is 0.9/1000. Interventions should be based on maternal/obstetric factors.
Equivocal BPP (perinatal mortality 7–10/1000)
• Score 8/10 with decreased AFV; in the absence of rupture of membranes this indicates an increased risk for chronic compensated hypoxaemia and/or acute-on chronic decompensation. Delivery is indicated in the presence of fetal lung maturity. For absent fetal lung maturity repeat testing daily.
• 6/10 with normal AFV (equivocal); indicates increased risk for acute asphyxia. In the presence of maturity delivery is indicated otherwise repeat testing in 24 hours should be performed.
Abnormal BPP (perinatal mortality 12–300/1000)
• 6/10 with decreased AFV; fetus is at risk for chronic asphyxia with possible acute asphyxia. In this group delivery is indicated if gestational age ˇ32 weeks. Repeat daily testing is warranted if gestational age is 5, and abnormal if either the NST is non-reactive or the AFI is 5 or less. A normal score gives similar reassurance as a normal five-component BPP. An abnormal modified BPP requires a full five-component evaluation to verify fetal compromise. Factors that affect the biophysical profile score
There are several important factors that can affect fetal dynamic variables and therefore interpretation of the BPP.
Absence of fetal breathing can occur in the maternal fasting state, and therefore may require retesting after a meal.
A decrease in amniotic fluid volume requires exclusion of membrane rupture.
Administration of corticosteroids (dexamethasone or bethamethasone) to promote fetal lung maturity for anticipated preterm delivery cause a transitory decline in fetal breathing, heart rate reactivity, variability, and fetal movements in the 48 hours following administration.
A reduction in amniotic fluid volume may be observed after 72 hours of administration.Administration of magnesium sulphate can result in decreased fetal activity due to the neuromuscular effects.
In fetal anomalies of the central nervous system, behaviour and heart rate variables may be abnormal due to effects on the central regulatory centres or the connecting pathways.
In certain fetal conditions, such as placenta-based growth restriction or twin–twin transfusion, the rate of clinical progression cannot be anticipated by the BPP alone and other testing modalities such as fetal Doppler are required to adjust surveillance intervals.
The interpretation of the BPP and the appropriate management steps therefore always require consideration of the clinical circumstances. Further reading
Baschat AA Integrated fetal testing in growth restriction: combining mutivessel Doppler and biophysical parameters. Ultrasound Obstet Gynecol 2003:21:1–8.
Carlan SJ, O’Brien WF. The effect of magnesium sulfate on the biophysical profile of normal term fetuses. Obstet Gynecol 1991;77:681–4.
Jackson JR, Kleeman S, Doerzbacher M, Lambers DS. The effect of glucocorticosteroid administration on fetal movements and biophysical profile score in normal pregnancies. J Matern Fetal Neonatal Med 2003;13:50–3.
Manning FA, Lange IR, Morrison I, Harman CR. Fetal biophysical profile score and non-stress test: a comparative trial. Obst Gynecol 1984;64:326–31.
Manning FA. Fetal biophysical profile. Obst Gynecol Clin North 1999;26:558.
Manning FA. Fetal biophysical profile: a critical appraisal. Clin Obst Gynecol 2002;45:975.
Cardiotocography Definition
Cardiotocography (CTG) is a well-established and widely practised method of fetal surveillance during labour. Its use has been extended to the antenatal period, and is often also referred to as the non-stress test (NST).
The aim of antenatal CTG is to screen and identify babies with acute/chronic hypoxia or those at risk of developing hypoxia.
Fetal hypoxia results in adaptations in the fetus that result in changes in heart rate patterns. Therefore CTG has become a screening tool in high-risk pregnancies (Table 7.2.1).At present CTG is not recommended as a method of routine fetal assessment in low-risk pregnancies in the UK. The technique
The CTG is a record of the fetal heart rate (FHR) obtained through a transducer placed on maternal abdomen, usually paired with another transducer that registers uterine activity. The registration is printed on a paper strip. Information regarding baseline fetal heart rate, variability, accelerations, and decelerations is provided.
Antenatal CTG is most commonly performed in the third trimester. But at times, can be performed early on but usually not before 26 weeks. A 20–30 minute long recording of the fetal heart rate pattern is often used on its own (the NST). Interpretation of the test
Normal/reassuring/reactive (Fig. 7.2.1)
• Should have at least two accelerations (>15 bpm for >15 seconds) in 20 minutes, baseline heart rate 110–160 bpm, baseline variability 5–25 bpm, absence of decelerations
Table 7.2.1 Indications for antenatal fetal monitoring

• Sporadic decelerations amplitude bpm
• Reduced baseline variability (5–10 bpm for >40 minutes)
• Baseline variability >25 bpm in the absence of accelerations
• Sporadic decelerations of any type unless severe as described below.

Fig 7.2.1 Normal antenatal cardiotocographs: normal rate, baseline variability, and accelerations.
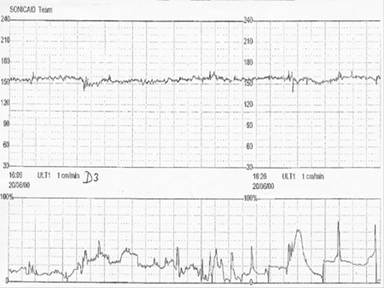
Fig 7.2.2 Equivocal antenatal cardiotocograph. There are no accelerations, the variability is reduced but no decelerations.
Pathological/ominous
• Baseline heart rate 180 bpm
• Silent pattern (baseline variability 40 minutes
• Sinusoidal pattern (oscillation frequency 10 bpm for >40 minutes with no accelerations and no period of normal baseline variability)
• Repeated late, prolonged (>1 minute) and severe variable (>40 bpm) decelerations (Fig.
7.2.3).The main drawback of antenatal CTG is that the analysis of the trace is visual, and there is lack of consistency even between ‘experts’. Indeed, the same trace is not interpreted consistently by the same observer.
The NST has been used in combination with other antenatal assessment tools as:
• Contraction stress test (CST): it is based on the intrapartum observation that linked recurrent late FHR decelerations occur with fetal hypoxaemia. The underlying mechanism for this event is a slowing of the fetal heart rate in response to transient systemic hypertension, provoked by reduction in arterial oxygen levels. As developed by Freeman, the CST was performed with intravenous oxytocin infusion until at least three moderate or strong contractions per 10 minutes were generated in a 20-minute window.
• The test was classified as follows:
• Negative: No late or significant variable decelerations.
• Positive: Late decelerations with at least 50% of contractions.
• Suspicious: Intermittent late or variable decelerations.
• This test has shown to have very low rate of false-negative results and false-positive rates at approximately 30%. In current practice, it is used very infrequently following an abnormal NST, due to the introduction of other non-invasive tests such as the biophysical profile (BPP), and Doppler velocimetry.
• Biophysical profile (BPP): assessment and scoring that includes fetal movements, fetal breathing movements, tone, amniotic fluid volume and assessment of FHR (Chapter 7.1). It was developed by Manning et al., based on the observation that fetal responses to hypoxia are not random, but occur in a precise order. FHR and breathing are affected first, followed by fetal movements and finally tone. Amniotic fluid measurement is important component of fetal biophysical profile.
• Doppler assessment: see chapter on Doppler Ultrasound.
• Computerized CTG: Is an automated evaluation of the fetal heart rate trace. There is a system of analysis that gives criteria of normality for computerized CTG known as Dawes/Redman criteria. The main advantage of a computerized system over visual interpretation is consistency in the interpretation of the CTG trace on different occasions.
Several studies suggest that among all information provided by computerized CTG, the most valuable in predicting fetal hypoxia is short-term variability (STV). Values of STV care: routine care for the healthy pregnant woman. London: RCOG Press 2008.
Turan S, Miller J, Baschat AA. Integrated testing and management in fetal growth restriction. Semin Perinatol 2008;32:194–200.

Fig. 7.2.5 Abnormal Doppler pathway. FM, fetal movements; IUD, intrauterine death; MCA, middle cerebral artery; SGA, small for gestational age; PI, pulsatility index.
Doppler ultrasound
Doppler ultrasound is now widely used to investigate the fetal circulation. ‘Fetal Doppler’ examination involves, in the vast majority of cases, evaluation of the umbilical and middle cerebral artery and, where indicated, the ductus venosus.
The commonest indication for fetal Doppler examination is in the context of a growth-restricted or compromised baby, where there might be fetal hypoxia or even acidaemia. In this context, fetal Doppler changes of individual vessels, using pulse wave Doppler, are highly correlated with hypoxia, and examination of the fetal circulation can give a clinically helpful assessment of the baby’s state of health. A recent development of fetal Doppler, the middle cerebral artery peak systolic velocity in the assessment of fetal anaemia, has made a major impact into the fetal medicine care of these babies.
This chapter considers these most common applications of fetal Doppler, and the theory underlying the practice. Types of Doppler and their clinical application
Most Doppler methods measure velocity in the direction of the ultrasound beam from which the colour flow and Doppler spectrum displays are produced. The requirements of colour flow imaging (including power Doppler) and pulsed wave Doppler are very different.
Colour flow imaging
Produces a colour map of flow over a region of the image. There is limited flow information—the colour shows the mean velocity vector at each point. In general, there is poor temporal resolution—because of the need to sample over a large area, flow images are usually updated at a low frame rate and only moderate spatial resolution.
Pulsed wave (spectral) Doppler
Examines flow at one point within a vessel and represents the distribution of flow velocities within the sample volume with time as the x axis. The static image allows calculation of velocity and flow waveform indices.
Power Doppler (also described as Doppler energy)
This is similar to colour flow imaging but directional information is sacrificed and temporal resolution is reduced to gain better sensitivity to low flow and low velocity situations. It is usually considered a qualitative technique, although there are methods to allow semi-quantitative analysis.
In the majority of applications for obstetric Doppler, a colour flow box is placed over the region of interest, then a specific blood vessel is visualized and then insonated using pulsed wave Doppler. These two Doppler modes will be discussed in more detail. Doppler energy/power Doppler can allow visualization of low velocity flow areas of the fetal circulation but provides mainly qualitative rather than quantitative information.
Fetal Doppler using colour flow imaging can be useful in assessing fetal anatomy; for example, intracardiac Doppler, which requires specialized Doppler settings; arteriovenous malformations; or the extent of liver displacement in diaphragmatic hernia. Basic physics
For pulsed wave and colour flow Doppler imaging, measurement of velocity is achieved by measurement of the change in phase in the returning echoes from blood at a particular time after transmission. This produces a Doppler frequency described in the well-known equation (Eqn 7.3.1):

Where:
ft is the transmitted ultrasound frequency,
V is the velocity of the blood
θ is the angle between the beam and the direction of flow
c is the speed of sound in tissue
The Doppler frequency determines the colour flow signal or Doppler spectrum. As the equation shows us, the Doppler frequency is dependent on
• blood velocity: as velocity increases so does the Doppler frequency;
• the angle between the Doppler pulse beam and the vessel: The Doppler frequency increases as the beam is at a narrower angle to the flow. There is very little or no signal at angles close to 90 degrees, which is often represented by no colour showing on the colour flow map at these angles.
• ultrasound frequency: higher ultrasound frequencies give increased Doppler frequencies. The ‘trade off’ is that lower ultrasound frequencies penetrate tissue better. How are the waveforms assessed and measured?
Multiple indices have been described for the assessment of resistance, for example A/B ratio; S/D ratio; (resistive or Pourcelot index (RI)); (pulsatility index (PI)). Similarly, velocity is described in many different ways: time-averaged velocity (TAV); time-averaged maximum velocity (TAMX); means velocity; peak systolic velocity (PSV); minimum velocity (Vmin), etc.
The most commonly used index, for which most charts exist for fetal Doppler, is the PI, and this index will be discussed below. For velocity, excluding blood flow measurements, PSV is almost universally used in the context of assessing fetal anaemia in the middle cerebral artery (Table 7.3.1). Fetal vessels
The umbilical artery
The umbilical artery waveform represents placental, not fetal, vascular resistance and should therefore be regarded primarily as an indicator of resistance in the fetoplacental vascular bed rather than representing the fetal circulation.
Studies from the late 1980s and early 1990s established the relationship between abnormal umbilical artery findings (absent end diastolic flow (EDF) or reversed EDF) and adverse perinatal outcome in the context of fetal growth restriction. A meta-analysis of studies of umbilical artery Doppler in a high-risk population showed improved perinatal outcomes where umbilical artery Doppler is performed, although the reason for this is not clear. More recently, it has been suggested that the risk for adverse outcome is highest in small babies with abnormal umbilical artery Doppler, but also appreciable in those with normal umbilical artery Doppler.
Table 7.3.1

How to perform umbilical artery Doppler
Umbilical artery waveforms are usually obtained from a free loop of umbilical cord, in most cases near the placental insertion where movement artefact is less. The angle of insonation should be less than 60 degrees. There is no consistent significant difference in the shape of the waveform depending upon where the cord is insonated, nor is it common for there to be a difference in waveform between the two arteries, although impedance indices are slightly higher at the fetal end of the umbilical cord, and lower at the placental insertion.
Three major abnormalities of umbilical artery flow are described:
• raised resistance (PI >95th centile)
• absent EDF
• reversed EDF.
From the sixteenth week onwards, the umbilical artery waveform should show positive end diastolic flow. Reduction in end diastolic flow, a rise in PI, absent EDF (Fig. 7.3.2), and reversed EDF represent increasing fetoplacental resistance. At 24–34 weeks, a fetus may have absent EDF in the umbilical artery for days or weeks before delivery is necessary. At very early gestations (between 24–32 weeks), even reversed EDF in the umbilical artery should not, without corroborating evidence from other fetal Doppler measurements, be the sole indication for delivery. At 32–34 weeks, delivery decisions in a growth-restricted baby may be made on the basis of amniotic fluid volume, movements, cardiotocography (CTG), and umbilical artery Doppler. After 34 weeks, absent EDF is unusual and almost always suggests severe fetoplacental pathology warranting delivery; at 32 weeks, reversed EDF would normally warrant delivery. Key points
• Umbilical artery Doppler is not the same as ‘fetal Doppler’, but gives an indication about fetoplacental vascular resistance. This does not necessarily correlate Doppler findings of the fetal circulation.
• Absent or reversed umbilical artery end diastolic flow should not dictate delivery prior to 32–34 weeks. This finding normally warrants more detailed investigation in units where fetal Doppler is available, or close observation and investigation using CTG in units where it is not. The middle cerebral artery
Examination of the fetal middle cerebral artery (MCA) relies on the physiological fetal adaptation to hypoxia called ‘brainsparing’ or ‘cerebral redistribution’. The normal, healthy MCA waveform shows little or no EDF, or even a little reverse flow. From 28–34 weeks end diastolic flow is often seen, and after 34 weeks, the MCA PI may be reduced to its maximum from ‘physiological redistribution’ due to changes in flow through the heart leading to relatively deoxygenated blood being shunted to the cerebral circulation.
In hypoxia there is a progressive reduction in resistance in the MCA (brain sparing). Where severe hypoxia leads to fetal acidaemia (fetal decompensation), the fetal MCA PI may however show a paradoxical increase in resistance for 24–48 hours before irreversible fetal heart rate changes or fetal death occur. Recent work suggests that hypoxia leading to cerebral redistribution involves subtle differences in perfusion of different areas of the fetal brain. Using a semi-quantitative technique known as fractional moving blood volume (FMBV), based on colour Doppler energy/power Doppler, perfusion of the hindbrain, forebrain, and midbrain has been shown to map differently in normal versus hypoxic fetuses. The clinical importance of this finding is as yet unknown.
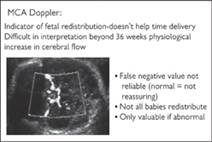
Fig. 7.3.1 Key points for performance of middle cerebral artery Doppler.
How to perform MCA Doppler
The fetal head is visualized in the biparietal diameter section, and the probe tilted to allow visualization of the greater wing of the sphenoid bone. The course of the MCA follows the wing of the sphenoid bone, allowing it to be seen easily on colour flow Doppler. The anterior vessel is insonated with pulsed wave Doppler in the segment nearest the Circle of Willis (Fig. 7.3.1). The MCA in anaemia
Measuring MCA peak systolic velocity (PSV) is useful for non-invasive monitoring of babies at risk of anaemia (for example rhesus disease) or a hyperdynamic circulation (for example sacrococcygeal teratoma). The technique is simple and reproducible; the MCA PSV correlates well with fetal anaemia: above the 1.5 multiples of the median (MoM) PSV for gestation, a baby is likely to have anaemia whereas below this level anaemia is very unlikely. Babies at risk of anaemia are frequently followed up on a weekly or 2 weekly basis in fetal medicine units, whereas previously cordocentesis was the only way to establish whether they were anaemic and required transfusion. The thoracic aorta
The thoracic aorta (TA) is the least frequently visualized major vessel in the investigation of fetal condition. Its flow velocity waveform mirrors the umbilical artery closely, although the resistance in it is usually higher.
Key points
• Fetal Doppler may become abnormal even in normally grown babies. This may occur with rapid onset pre-eclampsia or poorly controlled diabetes.
• Fetal Doppler may be normal after 34 weeks in compromised babies. Fetal Doppler assessment is not normally considered a reliable in the assessment of post-34-week pregnancies. Umbilical artery Doppler rarely shows major changes, and the MCA PI range already shows maximum physiological dilatation. Fetal venous Doppler
Umbilical vein
The umbilical vein can give important information especially if for technical reasons the fetal ductus venosus cannot be visualized. It normally shows a low velocity continuous flow. Using pulsed wave Doppler, it may be obtained transposed on an umbilical artery waveform; however, this is not recommended as reversed umbilical EDF may be ‘lost’ in the venous waveform in the opposite channel. Umbilical vein pulsations may be confused with the common physiological undulations associated with fetal breathing or even fetal movements.
Ductus venosus
The ductus venosus is a short, narrow connection between the umbilical vein and the right atrium of the heart. Early studies established the normal ranges for ductus venosus PI and characterized the changes associated with hypoxia and acidaemia. More recently, the ductus venosus waveform has been considered as the vessel most likely to differentiate between normal and abnormal outcome. ‘Early’ and ‘late’ changes in the ductus venosus waveform form the basis for the multicentre TRUFFLE randomized study (www.trufflestudy.org), a management study of severe fetal growth restriction.
How to perform ductus venosus Doppler
Oxygenated blood is directed from the umbilical vein into the fetal circulation towards the foramen ovale. The blood flow in the ductus venosus therefore reflects the pressure gradient between these two stuctures.
The ductus venosus waveform is quite distinguishable from that of the inferior vena cava and hepatic vein. The typical ductus venosus waveform has ‘s’, ‘d’, and ‘a’ waves.
The ductus venosus abnormalities are categorized below:
• raised ductus resistance (pulsatility index for vein >95th centile)
• exaggerated a wave approaching the baseline
• reversed a wave (a wave beneath the baseline).
A reversed a wave is an ominous sign and suggests fetal decompensation in the context of uteroplacental insufficiency severe hypoxia/acidaemia. This may also occur in the recipient twin in twin-to-twin transfusion syndrome, and in end-stage fetal anaemia or viral myocarditis. Integrating fetal Doppler into obstetric practice for the hypoxic baby
Fetal Doppler is useful tool in the management of fetal compromise, particularly in the context of placental insufficiency and hypoxia, and there is good correlation between hypoxia and impedance in individual fetal blood vessels. There are, however, few clues from the literature as to how fetal Doppler measurements should be integrated with assessment of fetal growth, amniotic fluid, movements, and cardiotocography in the management of a fetus. The Growth Restriction Intervention Trial (GRIT), which randomized ‘compromised’ babies into immediate or delayed delivery, did not show any significant difference in outcome between the two groups nor were there any clues in the use of Doppler. The TRUFFLE study (above) seeks to establish whether delivery of growth-restricted fetuses on the basis of abnormal CTG, ‘early’, or ‘late’ ductus venosus changes leads to an improved 2-year perinatal outcome; however, the results will not be available until 2012.
In this context, ‘pragmatic’ guidelines, such as those in Fig. 7.3.2, agreed by many perinatologists are reasonable to use in everyday practice until definitive evidence becomes available.

Fig. 7.3.2 Pragmatic consensus of delivery criteria in interuaterin growth restriction. Further reading
The GRIT study group. Infant wellbeing at 2 years of age in the Growth Restriction Intervention Trial (GRIT): multicentred randomised controlled trial. Lancet 2004;364:513–20.
Figueras F, Eixarch E, Gratacos E, Gardosi J. Predictiveness of antental umbilical artery Doppler for adverse pregnancy outcome in small-for-gestational-age babies according to customised birthweight centiles: population-based study. Br J Obstet Gynaecol 2008;115:590–4.
Loughna P. Intra-uterine growth restriction: investigation and management. Curr Obstet Gynaecol 2003;13:205–11.
Baschat AA, Galan HL, Bhide A, et al. Doppler and biophysical assessment in growth restricted fetuses: distribution of test results. Ultrasound Obstet Gynecol 2006;27:41–7.
Baschat AA. Doppler application in the deliver timing of the preterm growth-restricted fetus: another step in the right direction. Ultrasound Obstet Gynecol 2004;23:111–18.
Turan OM, Turan S, Gungor S, et al. Progression of Doppler abnormalities in intrauterine growth restriction. Ultrasound Obstet Gynecol 2008;32:160–7.
Figueras F, Benavides A, Del Rio M, et al. Monitoring of fetuses with intrauterine growth restriction: longitudinal changes in ductus venosus and aortic isthmus flow. Ultrasound Obstet Gynecol 2009;33:39–43.
Baschat AA, Viscardi RM, Hussey-Gardner B, et al. Infant neurodevelopment following fetal growth restriction: relationship with antepartum surveillance parameters. Ultrasound Obstet Gynecol 2009;33:44–50.
Tchirikov M, Schrãder HJ, Hecher K. Ductus venosus shunting in the fetal circulation: regulatory mechanism, diagnostic methods and medical importance. Ultrasound Obstet Gynecol 2006;27:452–61.
Figureras F, Fernandez S, Eixarch E, et al. Middle cerebral artery pulsatility index: reliability at different sampling sites. Ultrasound Obstet Gynecol 2006;28:809–13.
Figueras-Diesel H, Hernandez-Andrade E, Acosta-Rochas R, et al. Doppler changes in the main fetal brain arteries at different stages of hemodynamic adaptation in severe growth restriction. Ultrasound Obstet Gynecol 2007;30:297–302.
Hernandez-Andrade E, Figueroa-Diesel H, Janssons T, et al. Changes in the regional fetal cerebral blood flow perfusion in relation to hemodynamic deterioration in severely growth-restricted fetuses. Ultrasound Obstet Gynecol 2008;32:71–6.
Fetal abnormalities: cardiovascular Definition
Congenital heart disease (CHD) is defined as an abnormality of the cardiovascular system that is present at birth. The heart is a complex organ, but already formed at around 8 weeks of gestation. Most CHD includes structural defects, but abnormalities with primary myocardial involvement (e.g. cardiomyopathy) or rhythm disturbances (e.g. tachycardias and heart block) may also be congenital. Major heart defects can be defined as those that are lethal or require intervention, either surgical or by cardiac catheterization, in infancy or on long-term follow-up. Epidemiology
Structural CHD is one of the most serious forms of congenital defects and accounts for approximately 8 in 1000 live births with approximately half being considered major. The percentage of major CHD seen in fetal life is higher, as well as associated chromosomal defects, genetic syndromes, and extracardiac anomalies. Minor defects are being increasingly detected postnatally and if all minor problems are included, postnatal incidence may be as high as 5%. Aetiology and associations
Chromosomal, genetic abnormalities and environmental agents are well-recognized factors in the aetiology of CHD. Specific genes are increasingly being linked to CHD but non-specific factors and random errors may also lead to heart defects. A non-exhaustive list of some important associations is presented below.
Chromosomal defects and genetic syndromes
• Trisomy 21: About 40–50% of these fetuses will have CHD. The most characteristic defects are atrioventricular septal defect and perimembranous ventricular septal defect (VSD).
• Trisomy 13 and 18: Approximately 90% will have CHD, from simple septal defects to complex lesions. Large perimembranous inlet VSDs are commonly seen in trisomy 18 fetuses.
• Turner syndrome (monosomy X): Left heart involvement including coarctation of the aorta and hypoplastic left heart syndrome.
• Di George sequence and velocardiofacial syndrome (microdeletion of 22q11 and 10p deletion): Typically, conotruncal malformations such as tetralogy of Fallot, truncus arteriosus and interrupted aortic arch.
• Noonan syndrome (chromosome 12q): Pulmonary valve stenosis and hypertrophic cardiomyopathy.
• William syndrome (microdeletion 7q11): Supravalvar aortic stenosis and peripheral pulmonary artery stenosis.
• Holt–Oram syndrome (chromosome 12p): atrial septal defects.
• Ellis–van Creveld syndrome (chromosome 4p).
• VACTER association.
• CHARGE association.
Environmental and maternal factors
• Maternal infection (e.g. rubella)
• Maternal use of teratogenic drugs (e.g. lithium, antiepileptic medication, retinoic acid, alcohol)
• Maternal diabetes: about 2–3% risk of structural CHD in pregestational diabetes. Fetuses may also develop diabetic (hypertrophic) cardiomyopathy later in pregnancy.
• Maternal phenylketonuria
• Maternal collagen disorders (lupus and Sjogren disease). The presence of maternal antibodies (anti-Ro/SSA and anti-La/SSB) is associated with a 2 to 7.5% risk of conduction abnormalities in the fetus (heart block), occurring from around 18 weeks of gestation with the greater risk at 22–24 weeks. Recurrence risk is increased further to 16%.
Family history
Most congenital heart defects occur in low-risk pregnancies, but if there is an affected first-degree relative, the risk of CHD is increased. Table 7.4.1 gives an overall risk estimate for these families, but the relative risk may still vary according to the type of defect present in the proband.
Nuchal translucency
There is a clear association between increased nuchal translucency (NT) in the first trimester and the presence of major CHD. In chromosomally normal fetuses, the higher the NT thickness, the higher the risk of CDH. Table 7.4.1 gives a breakdown of risks depending on NT thickness. Natural history and antenatal prognosis
Spontaneous fetal demise due to CHD is relatively rare.
In general, most forms of structural CHD are well tolerated during pregnancy and thus do not require fetal therapy. Patency of the three natural shunts in the fetal circulation (the foramen ovale, ductus arteriosus and ductus venosus) allows the circulation to ‘bypass’ critical lesions and therefore ensure that most fetuses are haemodynamically stable throughout gestation, even though the neonate may require intervention in the first few days of life.
However, structural lesions which are associated with significant atrioventricular or semilunar valve regurgitation or myocardial dysfunction, have a more guarded outlook, as this often coexists with cardiomegaly and heart failure (fetal hydrops). Hydrops may also develop in a fetus with persistent tachyarrhythmia or complete heart block. In the former, hydrops is reversible if the tachycardia is controlled prenatally. In the latter, hydrops is associated with a poor prognosis.
Table 7.4.1 Groups at increased risk for CHD

Fetal therapy
Maternal administration of anti-arrhythmic drugs or, less often, direct fetal therapy may be indicated in cases of persistent tachyarrhythmias or when there is circulatory compromise. A decision to treat the arrhythmia prenatally should be balanced against early delivery and postnatal treatment of the newborn.
Fetal intervention for structural CHD such as aortic stenosis was initially attempted long ago but with disappointing results. However, with recent technical advances and better ultrasound image resolution, balloon dilatation of either aortic or pulmonary valves has been performed in highly selected cases in an attempt to improve postnatal morbidity. If balloon valvulopasty of stenotic valves or atrial septectomy is to be performed in the fetus, a multi-disciplinary approach in specialist tertiary referral centres is essential. Long-term results are not available. Clinical approach to CHD in the fetus
Screening for CHD
Most screening programmes incorporate assessment of the four-chamber view at the time of the 18–23 week scan. Antenatal detection rates based on this alone are generally low, but there is wide regional variation. Screening programmes that include assessment of the outflow tracts have higher detection rates.
In the UK, recent guidelines of the National Institute for Clinical Excellence (NICE) and the Fetal Anomaly Screening Programme (FASP) recommended that all pregnancies be screened by a combination of four-chamber and outflow tract views.
Referral to tertiary centre
Fetal echocardiography should be offered to families at risk of CHD or when an abnormality is suspected.
Suspected (structural or functional) abnormality
If a fetal cardiac abnormality is suspected at any time during pregnancy (often at the routine 18–23 week scan) referral to a specialist in fetal echocardiography should be made as soon as possible. Specialist assessment is essential for accurate diagnosis of the abnormality and to allow concomitant or subsequent counselling by the fetal and/or paediatric cardiologist. A multidisciplinary team that also includes a specialist in fetal medicine and clinical genetics is indicated to assess the presence (or not) of extracardiac malformations and to perform invasive tests, if appropriate.
High-risk pregnancies
Families considered to be at higher risk of CHD should be referred for elective fetal echocardiography, around the time of the routine obstetric scan (18–23 weeks). In selected centres where there are experts in early fetal echocardiography, cardiac scans may be performed from around 12 weeks of gestation. This may be offered to fetuses with markedly increased NT (usually NT >4 mm) or to families with previous history of CHD (usually for reassurance). Owing to the relatively high number of associated noncardiac problems, a multidisciplinary team approach to early scans is highly desirable.
Ultrasound: diagnostic fetal echocardiography
The variable types of CHD, their wide morphological spectrum and, often, their complex nature means that accurate diagnosis requires the input of a professional who is highly familiar with congenital cardiac malformations and their manifestation both prenatally and postnatally. There are various terminologies used to describe CHD. The sequential segmental analysis to diagnosis offers a logical approach to describing simple and most importantly, complex malformations.
Counselling and pregnancy management
Extensive consultation usually follows the diagnosis of major CHD in the fetus. Postnatal management options and timing of intervention for the neonate/child with CHD varies with each diagnosis and will be provided by the paediatric/fetal cardiologist. Associated extracardiac malformations need to be diagnosed or excluded by an experienced fetal medicine specialist.
Consideration needs to be given to the option of invasive tests (CVS, amniocentesis or cordocentesis) to assess fetal karyotype. Depending on gestational age at the time of diagnosis, the severity of the lesion and family/religious/social and legal issues, the option of termination of pregnancy will also be discussed.
Follow-up cardiac scans are usually planned at a few weeks’ interval. Fetal growth may also be monitored. The importance of a multidisciplinary team approach cannot be overemphasized and a clear perinatal plan should be in place to ensure optimal clinical care of mother and baby. Perinatal management
The various forms of CHD may be broadly divided into the following categories regarding perinatal management:
CHD requiring early neonatal treatment
Duct-dependent lesions require elective intravenous infusion of prostaglandin E to maintain patency of the ductus arteriosus. This allows early transfer to a cardiac unit where surgery/intervention will be performed prior to discharge from hospital.
Duct-dependent systemic circulation
• Coarctation of the aorta/interrupted aortic arch
• Critical aortic stenosis
• Hypoplastic left heart syndrome
• Complex lesions associated with severe systemic outflow obstruction/aortic arch obstruction.
Duct-dependent pulmonary circulation
• Pulmonary atresia with VSD
• Pulmonary atresia/critical pulmonary stenosis with intact ventricular septum
• Complex lesions associated with critical pulmonary stenosis/atresia.
Simple, complete transposition of the great arteries Tachy- and bradyarrhythmias
Major CHD associated with neonatal stability
This group includes defects that have a balanced circulation at birth. The newborn baby is expected to be stable, albeit cyanosed in many instances. An elective postnatal assessment should be organized to plan further cardiac follow-up. Time of surgery or interventional cardiac catheterization is often beyond the first month of life.
• Septal defects without significant outflow tract obstruction.
• Tetralogy of Fallot with pulmonary stenosis and adequate forward flow.
• Complex lesions without significant/critical systemic or pulmonary outflow obstruction.
Relatively minor CHD that may not require any treatment
Follow-up can be organized for a few weeks after birth in order to document postnatal findings and plan further follow-up, if needed. Included in this group are small muscular VSDs. Place and timing of delivery
Delivery should take place where there are good neonatal facilities to support the needs of the newborn baby with CHD, including artificial ventilation if necessary. Women whose fetuses have complete transposition should have their obstetric care transferred to deliver as close as possible to the cardiac unit as balloon atrial septostomy may be required shortly after birth.
In general, delivery will take place at term, providing there are no concerns regarding fetal growth or progression of the cardiac disease to such an extent as to impact on postnatal management.
Most fetuses can be delivered vaginally, but induction of labour may be necessary in order to plan neonatal surgery. A Caesarean section is rarely indicated for cardiac reasons. Exceptions include rhythm abnormalities (e.g. heart block and tachyarrhythmias) that may impact on fetal heart rate monitoring during labour. Further reading
Lee W, Allan LD, Carvalho JS, et al. ISUOG Consensus Statement: What constitutes a fetal echocardiogram? Ultrasound Obstet Gynecol 2008;32:249–52.
Carvalho JS, Ho SY, Shinebourne EA. Sequential segmental analysis in complex fetal cardiac abnormalities: a logical approach to diagnosis. Ultrasound Obstet Gynecol 2005;26:105–111
Hyett JA, Perdu M, Sharland GK, et al. Increased nuchal translucency at 10–14 weeks of gestation as a marker for major cardiac defects. Ultrasound Obstet Gynecol 1997;10:242–46.
Ghi T, Huggon IC, Zosmer N, et al. Incidence of major structural cardiac defects associated with increased nuchal translucency but normal karyotype. Ultrasound Obstet Gynecol 2001;18:610–14.
Tegnander E, Eik-Nes SH, Johansen OJ, et al. Prenatal detection of heart defects at the routine fetal examination at 18 weeks in a non-selected population. Ultrasound Obstet Gynecol 1995;5:372–80.
Bull C. Current and potential impact of fetal diagnosis on prevalence and spectrum of serious congenital heart disease at term in the UK. Lancet 1999;354:1242–7.
Carvalho JS, Moscoso G, Tekay A, et al. Clinical impact of first and early second trimester fetal echocardiography on high risk pregnancies. Heart 2004;90:921–6.
Bonnet D, Coltri A, Butera G, et al. Detection of transposition of the great arteries in fetuses reduces neonatal morbidity and mortality. Circulation 1999;99:916–8.
Internet resources
National Institute for Clinical Excellence: www.nice.org.uk British Heart Foundation: www.bhf.org
Children’s Heart Federation: www.childrens-heart-fed.org.uk
Fetal abnormalities: central nervous system Neural tube defects
Definition
Neural tube defects include a group of anomalies that share failure of closure (or secondary reopening) of the neural tube: anencephaly, spina bifida and encephalocele.
Incidence
This is subject to large geographical and temporal variations; in the UK the prevalence is about 5 per 1000 births. Anencephaly and spina bifida, with an approximately equal prevalence, account for 95% of the cases, and encephalocele for the remaining 5%. About 90% of cases of spina bifida identified at birth are open.
Aetiology
Chromosomal abnormalities, single mutant genes and maternal diabetes mellitus, or ingestion of teratogens, such as antiepileptic drugs, are implicated in about 10% of the cases. However, the precise aetiology for the majority of these defects is unknown. When a parent or previous sibling has had a neural tube defect, the risk of recurrence is 5–10%. Periconceptual supplementation of the maternal diet with folate reduces by about half the risk of developing these defects.
Pathology
In anencephaly there is absence of the cranial vault (acrania) with secondary degeneration of the brain. Encephaloceles are cranial defects, usually occipital, with herniated fluidfilled or brain-filled cysts. In spina bifida the neural arch is incomplete. Spina bifida is subdivided into open and closed lesions. In open lesions the neural tube is exposed to the external environment and there is always a malformative process of the neural cord. In closed spina bifida the defect is covered by skin and the neural tube is usually intact, although it may undergo secondary damage because of adhesions or compression.
Clinical approach: diagnosis
Anencephaly is usually rapidly recognized from 11 weeks’ gestation. At this time the main finding is the absence of the calvarium with exposure of the cerebrum, that usually appears severely deformed (Fig. 7.5.1). This is the first stage of anencephaly, which is also frequently referred to as acrania, or exencephaly. In the following weeks there is progressive reabsorption of the abnormal brain tissue, which is usually completely absent by the second trimester. Associated spinal lesions are found in up to 50% of cases. In the first trimester the diagnosis can be made after 11 weeks, when ossification of the skull normally occurs.
Diagnosis of open spina bifida requires the systematic examination of each neural arch from the cervical to the sacral region both transversely and longitudinally. In the transverse scan the normal neural arch appears as a closed circle with an intact skin covering, whereas in spina bifida the arch is ‘U’ shaped and there is an associated bulging myelomeningocele (thin-walled sometimes septated cyst). The extent of the defect and any associated kyphoscoliosis are best assessed in the longitudinal scan (Fig. 7.5.2).
The diagnosis of open spina bifida has been greatly enhanced by the recognition of associated abnormalities in the skull and brain. These abnormalities include frontal bone scalloping (lemon sign), and obliteration of the cisterna magna with either an ‘absent’ cerebellum or abnormal anterior curvature of the cerebellar hemispheres (banana sign) (Fig. 7.5.2). These easily recognizable alterations in skull and brain morphology are often more readily attainable than detailed spinal views. A variable degree of ventricular enlargement is present in virtually all cases of open spina bifida at birth, but in only about 70% of cases in the midtrimester. In England and in many Western countries, screening for fetal neural tube defects with maternal serum alphafetoprotein (where women with an increased concentration of alphafetoprotein (usually 2.5 MoM or more) are referred for a detailed ultrasound examination) has largely been replaced by sonographic screening.
In about 10% of cases, spina bifida is closed, that is covered by skin. In these cases, fetal intracranial anatomy is normal.
Encephaloceles are recognized as cranial defects with herniated fluid-filled or brain-filled cysts. They are most commonly found in an occipital location (75% of the cases) but alternative sites include the frontoethmoidal and parietal regions.

Fig. 7.5.1 (a) anencephaly in late first trimester; the calvarium is absent and distorted brain tissue (arrow) is seen arising from the skull base and floating in the amniotic fluid; (b) cephalocele: severe ventriculomegaly associated with a posterior protrusion of intracranial contents (arrow).
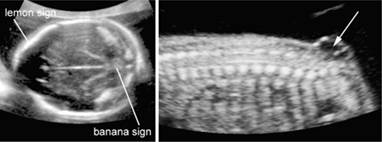
Fig. 7.5.2 (a) Arnold–Chiari malformation in a fetus with open spina bifida: there is frontal bossing (also referred to as lemon sign) the cerebellum is poorly delineated because of the absence of fluid in the cisterna magna (also referred to as banana sign); (b) myelomeningocele in a midtrimester fetus in the sacral area the neural canal is open and communicates with a septated cystic mass.
Prognosis
Anencephaly is fatal at or within hours of birth. In encephalocele the prognosis is inversely related to the amount of herniated cerebral tissue; overall, neonatal mortality is about 40% and more that 80% of survivors are intellectually and neurologically handicapped. In open spina bifida the surviving infants are often severely handicapped, with paralysis in the lower limbs and double incontinence; despite the associated hydrocephalus requiring surgery, intelligence may be normal. The outcome of closed spina bifida is difficult to predict. Some infants are completely asymptomatic. In other cases, neurological deficits including lower limb weakness to complete paralysis and urinary incontinence may be found.
Management
Termination of pregnancy can be offered to couples. In continuing pregnancies there is no indication to modify standard obstetric management. It has been debated whether fetuses with open spina bifida may benefit from Caesarean section, but no clear evidence exists.
Fetal therapy
There is some experimental evidence that in utero closure of spina bifida may reduce the risk of handicap because the amniotic fluid in the third trimester is thought to be neurotoxic. However, there are high risk from such intervention and it remains part of research.
Prevention
Periconceptual supplementation with folic acid reduces the risk of neural tube defects by as much as 50%. The recommended dosage is 400 μg daily for low-risk pregnancies and 4 mg in patients with an increased risk because of a positive familial history. Ventriculomegaly
Definition
Enlargement of the cerebral lateral ventricles.
Prevalence
Ventriculomegaly (lateral ventricle diameter of 10 mm or more) is found in 1% of pregnancies at the 20–23 week scan.
Aetiology
This may result from chromosomal and genetic abnormalities, intrauterine haemorrhage or congenital infection, although many cases have as yet no clear-cut aetiology.
Clinical approach: diagnosis
Fetal ventriculomegaly is diagnosed sonographically by the demonstration of abnormally dilated lateral cerebral ventricles (Fig. 7.5.3). Certainly before 24 weeks and particularly in cases of associated spina bifida, the head circumference may be small rather than large for gestation. A transverse scan of the fetal head at the level of the cavum septum pellucidum will demonstrate the dilated lateral ventricles, defined by a diameter of 10 mm or more of the posterior horn. The choroid plexuses, which normally fill the lateral ventricles are surrounded by fluid. Ventriculomegaly is commonly subdivided into two main groups: mild ventriculomegaly (10–15 mm) and severe ventriculomegaly (15 mm or more).

Fig. 7.5.3 Ventriculomegaly: the arrows indicate the distended lateral ventricles.
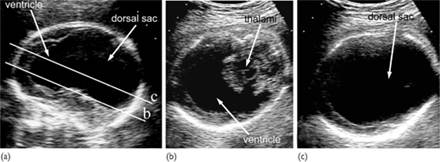
Fig. 7.5.4 Alobar holoprosencephaly in the midtrimester. (a) median plane demonstrating the single ventricular cavity, that has a rim of cortex anteriorly and amply communicates posteriorly with a dorsal sac; (b) axial scan at the level of the thalamus, demonstrating the crescent shaped single ventricle and the absence of the midline in the anterior cortex; (c) in a slightly craniad axial plane than the previous one, the communication between the ventricular cavity and the dorsal sac is demonstrated.
Prognosis
Fetal or perinatal death and neurodevelopment in survivors are strongly related to the presence of other malformations and chromosomal defects. Although mild ventriculomegaly (atrial width of 10–15 mm) is generally associated with a good prognosis, it is also the group with the highest incidence of chromosomal abnormalities (often trisomy 21). In addition, in a few cases with apparently isolated mild ventriculomegaly there may be an underlying cerebral maldevelopment (such as lissencephaly) or destructive lesion (such as periventricular leukomalacia). Recent evidence suggests that in about 10% of cases there is mild to moderate neurodevelopmental delay. Severe ventriculomegaly is associated with a much increased risk of neurological compromise, that is some studies is in the range of 50% of cases. Fetuses with severe ventriculomegaly may develop intracranial hypertension (hydrocephalus) and require postnatal drainage.
Fetal therapy
There is some experimental evidence that in utero cerebrospinal fluid diversion may be beneficial. However, attempts in the 1980s to treat hydrocephalic fetuses by ventriculo-amniotic shunting have now been abandoned because of poor results, mainly because of inappropriate selection of patients.
Management
Excluding associated anomalies is critical. This requires a detailed examination of cerebral and extracerebral anatomy with ultrasound. Fetal karyotyping and work-out for cytomegalovirus and toxoplasmosis infections should be offered. The use of magnetic resonance imaging has also been advocated, although the relative value of this examination compared with ultrasound remains controversial. In continuing pregnancies no modification of standard obstetric management is required. Caesarean section is only clearly indicated in cases with associated macrocrania. Holoprosencephaly
Definition
This is a spectrum of cerebral abnormalities resulting from incomplete separation of the forebrain. There are three types according to the degree of forebrain cleavage. The alobar type, which is the most severe, is characterized by a monoventricular cavity and fusion of the thalami. In the semilobar type there is partial segmentation of the ventricles and cerebral hemispheres posteriorly with incomplete fusion of the thalami. In lobar holoprosencephaly there is normal separation of the ventricles and thalami but absence of the septum pellucidum. The first two types are often accompanied by microcephaly and facial abnormalities.
Prevalence
Holoprosencephaly is found in about 1 in 10 000 births.
Aetiology
Although in many cases the cause is a chromosomal abnormality (usually trisomy 13) or a genetic disorder with an autosomal dominant or recessive mode of transmission, in many cases the aetiology is unknown. The risk of recurrence for sporadic, non-chromosomal holoprosencephaly, the empirical recurrence risk is 6%.
Clinical approach: diagnosis
In the standard transverse view of the fetal head for measurement of the biparietal diameter there is a single dilated midline ventricle replacing the two lateral ventricles or partial segmentation of the ventricles (Fig. 7.5.4). The alobar and semilobar types are often associated with facial defects, such as hypotelorism or cyclopia, facial cleft and nasal hypoplasia or proboscis.
Prognosis
Alobar and semilobar holoprosencephaly are lethal. Lobar holoprosencephaly is associated with mental retardation
Management
Given the poor prognosis of these defects, termination of pregnancy can be offered to the couples. In continuing pregnancies, no modification of standard obstetric management is indicated. Agenesis of the corpus callosum
Definition
The corpus callosum is a bundle of fibres that connects the two cerebral hemispheres. It develops at 12–18 weeks of gestation. Agenesis of the corpus callosum may be either complete or partial (usually affecting the posterior part).
Prevalence
Agenesis of the corpus callosum is found in about 5 per 1000 births.
Aetiology
Agenesis of the corpus callosum may be due to maldevelopment or secondary to a destructive lesion. It is commonly associated with chromosomal abnormalities (usually trisomies 18, 13 and 8) and more than 100 genetic syndromes.
Clinical approach: diagnosis
The corpus callosum is not visible in the standard transverse views of the brain but agenesis of the corpus callosum may be suspected by the absence of the cavum septum pellucidum and the ‘teardrop’ configuration of the lateral ventricles (enlargement of the posterior horns). Agenesis of the corpus callosum is demonstrated in the midcoronal and midsagital views, which may require vaginal sonography (Fig. 7.5.5).
Prognosis
This depends on the underlying cause. Prenatal studies indicate that 50–100% of fetuses with isolated agenesis of the corpus callosum will have a normal to borderline intelligence at long-term follow-up. Recent studies however suggest a progressive decline in intellectual capacity over the years.
Management
Excluding associated anomalies is critical. This requires a detailed examination of cerebral and extracerebral anatomy with ultrasound. Fetal karyotyping should be offered. The use of Magnetic resonance imaging has also been advocated, although the relative value of this examination compared with ultrasound remains controversial. In continuing pregnancies no modification of standard obstetric management is required. Dandy–Walker complex
Definition
The Dandy–Walker complex refers to a spectrum of cystic abnormalities of the cerebellum. Most cases diagnosed in utero will fall into one of these categories: (a) Dandy–Walker malformation (cystic dilatation of the fourth ventricle that occupies and distend the cisterna magna associated with superior rotation of the cerebellar vermis); (b) vermian hypoplasia (absence of part of the cerebellar vermis usually associated with a cystic dilatation of the fourth ventricle that does not distend the cisterna magna); (c) Blake’s pouch cyst (cystic dilatation of the fourth ventricle that causes a superior rotation of the cerebellar vernis that is intact, with a normal sized cisterna magna) and (d) mega-cisterna magna (large cisterna magna, normal vermis, and fourth ventricle).
Prevalence
Dandy–Walker malformation is found in about 1 per 30 000 births. No clear-cut epidemiological data exist with regard to the other entities.
Aetiology
The Dandy–Walker complex is a non-specific endpoint of chromosomal abnormalities (usually trisomies 18 or 13 and triploidy), genetic syndromes, congenital infection or teratogens such as warfarin, but it can also be an isolated finding.
Clinical approach: diagnosis
Enlarged cisterna magna is diagnosed if the vertical distance from the vermis to the inner border of the skull is more than 10 mm (Fig. 7.5.6). Dandy–Walker malformation, vermian hypoplasia and Blake’s pouch cyst share in common a cystic dilatation of the fourth ventricle and may be difficult to differentiate. In expert hands, careful scanning with multiple views may however identify the expansion of the cisterna magna with superior elevation of the sinus confluence that is typical of Dandy–Walker malformation, the incomplete formation of the vermis in the presence of a normal cisterna magna that is typical of vermian hypoplasia and the normal appearance of both cerebellum and cisterna magna that is typical of the Blake’s pouch cyst (Fig. 7.5.7).

Fig. 7.5.5 Complete agenesis of the corpus callosum; (a) axial plane: the frontal horns are more distant than normal, the cavum septi pellucidi is not present and in its position only a distended interhemispheric fissure is seen; there is a slight enlargement of the atria; the increased separation between the frontal horns and the enlargement of the atria result in a tear-shaped configuration of the ventricle; (b) coronal view: the frontal horns are more distant than normal and have a typical ‘comma’ shaped appearance; the interhemispheric fissure is distended and the two cerebral hemispheres are separated without any intervening corpus callosum; (c) sagittal view: above the area of the third ventricle the complex formed by the corpus callosum and cavum septi pellucidi is absent and replaced by the fluid contained into the interhemispheric fissure (3v, third ventricle; At, atria; FH, frontal horns, IHF interhemispheric fissure).
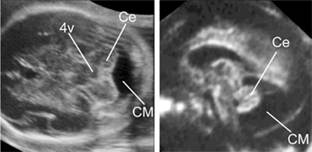
Fig. 7.5.6 Megacisterna magna in axial and sagittal views the cisterna magna is enlarged but the cerebellum appears intact (4v, fourth ventricle; Ce, cerebellum; CM, cisterna magna).
Prognosis
Prognosis depends heavily upon the presence of associated anomalies that are very frequently encountered. Blake’s pouch cyst and megacisetrna magna usually have a normal outcome when isolated, and intrauterine regression is often documented. The experience with Dandy–Walker malformation and vermian hypoplasia is limited and no clear-cut figures exist. It would seem however that isolated cases may be completely asymptomatic.
Management
Excluding associated anomalies is critical. This requires a detailed examination of cerebral and extracerebral anatomy with ultrasound. Fetal karyotyping should be offered. The use of magnetic resonance imaging has also been advocated, although the relative value of this examination compared with ultrasound remains controversial. In continuing pregnancies no modification of standard obstetric management is required. Microcephaly
Definition
Small head and brain.
Prevalence
Microcephaly is found in about 1 in 1000 births.
Aetiology
This may result from chromosomal and genetic abnormalities, fetal hypoxia, congenital infection and exposure to radiation or other teratogens, such maternal anticoagulation with warfarin. It is commonly found in the presence of other brain abnormalities, such as encephalocele or holoprosencephaly.
Clinical approach: diagnosis
The diagnosis is certain when the fetal head circumference is extremely small, 3 SD or more below the mean. However, in many cases the condition is progressive and diagnosis is not possible until late in gestation or after birth. The association with intracranial anomalies (roughly 50% of cases) greatly increases the index of suspicion.
Prognosis
This depends on the underlying cause, but in more than 50% of cases there is severe mental retardation.
Management
Fetal karyotyping and work-out of fetal infection should be offered. In continuing pregnancies no modification of standard obstetric management is required. Destructive cerebral lesions
Definition
These lesions include hydranencephaly, porencephaly and schizencephaly. In hydranencephaly, there is absence of the cerebral hemispheres with preservation of the midbrain and cerebellum. In porencephaly, there are cystic cavities within the brain that usually communicate with the ventricular system, the subarachnoid space or both. Schizencephaly is associated with clefts in the fetal brain connecting the lateral ventricles with the subarachnoid space.

Fig. 7.5.7 Differential diagnosis of open fourth ventricle; (a) open fourth ventricle in the axial view; (b) Dandy–Walker malformation: the sagittal view (is the most useful approach for a specific diagnosis; the posterior fossa is distended by a fluid accumulation; the cerebellar vermis (arrow) is rotated superiorly; hypoplasia is inferred by the small dimensions and by the absence of the common landmarks, the triangular shape of the fourth ventricle and the main fissures. Notice the high riding tentorium; (c) vermian hypoplasia; the cerebellar vermis (arrow) is rotated superiorly, is very small and is lacking the normal anatomic landmarks; (d) Blake’s pouch cyst the cerebellar vermis appears intact and is slightly rotated superiorly, with fluid interposed between it and the brain stem (arrow).
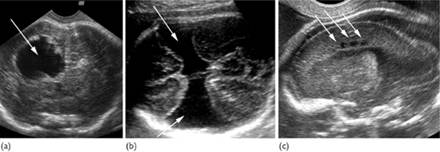
Fig. 7.5.8 Destructive lesions (arrows) of the fetal brain: (a) porencephalic cyst; (b) schizencephaly; (c) periventricular leucomalacia.
Prevalence
Destructive cerebral lesions are found in about 1 in 10 000 births.
Aetiology
Hydranencephaly is a sporadic abnormality that may result from widespread vascular occlusion in the distribution of the internal carotid arteries, prolonged severe hydrocephalus, or an overwhelming infection such as toxoplasmosis or cytomegalovirus. Porencephaly may be caused by infarction of the cerebral arteries or haemorrhage into the brain parenchyma. Schizencephaly may be a primary disorder of brain development or it may be due to early bilateral occlusion of the middle cerebral arteries.
Clinical approach: diagnosis
Differentiation between hydranencephaly and severe hydrocephalus may be difficult at times: the former condition should be suspected when no cerebral mantle can be demonstrated; even with the most severe form of hydrocephalus, a thin cortex and a midline echo are usually demonstrated. In porencephaly there is one or more cystic area in the cerebral cortex, which usually communicates with the ventricle (Fig. 7.5.8); the differential diagnosis is from intracranial cysts (arachnoid, glyoependymal) that are usually found either within the scissures or in the midline and compress the brain. In schizencephaly there are bilateral clefts extending from the lateral ventricles to the subarachnoid space, this is usually associated with absence of the cavum septum pellucidum (Fig. 7.5.8).
Prognosis
Hydranencephaly is usually incompatible with survival beyond early infancy. The prognosis in porencephaly is related to the size and location of the lesion and although there is increased risk of impaired neurodevelopment in some cases development is normal. Schizencephaly is usually associated with severe neurodevelopmental delay and seizures. Further reading
Adamsbaum C, Moutard ML, Andre C, et al. MRI of the fetal posterior fossa. Pediatr Radiol 2005;35:124–40.
Bennett GL, Bromley B, Benacerraf BR. Agenesis of the corpus callosum: prenatal detection usually is not possible before 22 weeks of gestation. Radiology 1996;199:447–50.
Blaas HG, Eik-Nes SH, Vainio T, Isaksen CV. Alobar holoprosencephaly at 9 weeks gestational age visualized by two- and three-dimensional ultrasound. Ultrasound Obstet Gynecol 2000;62–5.
Blaas HG, Eriksson AG, Salvesen KA, et al. Brains and faces in holoprosencephaly: pre- and postnatal description of 30 cases. Ultrasound Obstet Gynecol. 2002;19:24–38.
Boddaert N, Klein O, Ferguson N, et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology 2003;320–4.
Boyd PA, Wellesley DG, De Walle HE, et al. Evaluation of the prenatal diagnosis of neural tube defects by fetal ultrasonographic examination in different centres across Europe. J Med Screen 2000;7:169–74.
Bromley B, Benacerraf BR. Difficulties in the prenatal diagnosis of microcephaly. J Ultrasound Med. 1995;303–6.
Chervenak FA, Jeanty P, Cantraine F, et al. Spina bifida and anencephaly before and after folic acid mandate–United States, 199 996 and 199 000. MMWR Morb Mortal Wkly Rep. 2004;53:362–5.
Chervenak FA, Rosenberg J, Brightman RC, et al. A prospective study of the accuracy of ultrasound in predicting fetal microcephaly. Obstet Gynecol. 1987;69:908–10.
Filly RA, Cardoza JD, Goldstein RB, Barkovich AJ. Detection of fetal central nervous system anomalies: a practical level of effort for a routine sonogram. Radiology 1989;403–8.
Pilu G. Sonographic demonstration of brain injury in fetuses with severe red blood cell alloimmunization undergoing intrauterine transfusions. Ultrasound Obstet Gynecol 2004;23:428–31.
Ghi T, Brondelli L, Simonazzi G, et al. Outcome of antenatally diagnosed intracranial hemorrhage: case series and review of the literature. Ultrasound Obstet Gynecol 2003;22:121–30.
Guibaud L, des Portes V. Plea for an anatomical approach to abnormalities of the posterior fossa in prenatal diagnosis. Ultrasound Obstet Gynecol 2006;27:477–81.
Gupta JK, Bryce FC, Lilford RJ. Management of apparently isolated fetal ventriculomegaly. Obstet Gynecol Surv. 1994;49:716–21. Gupta JK, Lilford RJ. Assessment and management of fetal agenesis of the corpus callosum. Prenat Diagn 1995;15:301–12.
Hobbins JC. The diagnosis of fetal microcephaly. Am J Obstet Gynecol 1984;512–7.
Johnson SP, Sebire NJ, Snijders RJ, et al. Ultrasound screening for anencephaly at 11–14 weeks of gestation. Ultrasound Obstet Gynecol 1997;9:14–6.
Klein O, Pierre-Kahn A, Boddaert N, et al. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst 2003;19:484–9.
Malinger G, Lev D, Kidron D, et al. Differential diagnosis in fetuses with absent septum pellucidum. Ultrasound Obstet Gynecol 2005;25:42–9.
Malinger G, Lev D, Zahalka N, et al. Fetal cytomegalovirus infection of the brain: the spectrum of sonographic findings. AJNR Am J Neuroradiol 2003;24:28–32.
Melchiorre K, Bhide A, Gika AD, Pilu G. Papageorghiou AT. Ultrasound Obstet Gynecol 2009;34:212–24.
Moutard ML, Kieffer V, Feingold J, et al. Agenesis of corpus callosum: prenatal diagnosis and prognosis. Childs Nerv Syst 2003;19:471–6.
Nicolaides KH, Campbell S, Gabbe SG, Guidetti R. Ultrasound screening for spina bifida: cranial and cerebellar signs. Lancet 1986;2:72–4.
Pilu G, Falco P, Perolo A, et al. Differential diagnosis and outcome of fetal intracranial hypoechoic lesions: report of 21 cases. Ultrasound Obstet Gynecol 1997;9:229–36.
Pilu G, Sandri F, Perolo A, et al. Sonography of fetal agenesis of the corpus callosum: a survey of 35 cases. Ultrasound Obstet Gynecol 1993;3(5):318–29.
Volpe P, Paladini D, Resta M, et al. Characteristics, associations and outcome of partial agenesis of the corpus callosum in the fetus. Ultrasound Obstet Gynecol 2006;27:509–16.
Zalel Y, Gilboa Y, Gabis L, et al. Rotation of the vermis as a cause of enlarged cisterna magna on prenatal imaging. Ultrasound Obstet Gynecol 2006;27:490–3.
Fetal abnormalities: chromosomal anomalies Definition
An abnormality in the number or structure of one or more chromosomes. Epidemiology
The frequency of chromosomal abnormalities is dependent on the age distribution of the population in question as aneuploidies are age related. Pathology
Abnormal chromosome results
Down’s (trisomy 21), Edward’s (trisomy 18) and Patau’s (trisomy 13) are serious well-described abnormalities associated with severe mental handicap and multiple other congenital abnormalities. Survival even in the absence of major structural malformations in trisomies 13 and 18 is extremely limited; the median survival is 10–15 days. Expectant management during labour is appropriate following discussion with the parents in those that continue the pregnancy.
Fetal aneuploidy: autosomal aneuploidy
Down’s syndrome
↑ Nuchal translucency at 11–13 + 6 weeks; also congenital heart disease especially atrioventricular septal defect (AVSD), hyperechogenic bowel, duodenal atresia, other features very subtle.
Edward’s syndrome
↑ Nuchal Translucency at 11–13 + 6 weeks; also exomphalos, megacystis, strawberry-shaped head, multiple choroid plexus cysts, congenital heart disease (VSD, dysplastic valves), rocker bottom feet, renal abnormalities, intrauterine growth restriction (IUGR).
Patau syndrome
↑ Nuchal translucency at 11–13 + 6 weeks; also megacystis, exomphalos, holprosencephaly, tachycardia, cleft lip and palate, anophthalmia, polydactyly, congenital heart disease, renal cystic disease.
Triploidy 69,XXY or XXX
Severe early onset symmetrical IUGR. Redating has often occurred at the first trimester scan. Otherwise there are inconsistent ultrasound features such as ↑ nuchal translucency at 11–13 + 6 weeks; spina bifida, congenital heart disease, risk of severe early-onset preeclampsia, abnormal placenta.
Sex chromosome aneuploidies
Turner’s syndrome 45,X0
• Very high NT (lethal form), increasing lethality with increasing NT.
• NT all the important genetic material is present) (Fig. 7.6.1). Incidence 1:1000.
If it involves chromosomes 14 or 15 check that the fetus has inherited one copy of each chromosome from each parent (UPD, uniparental disomy) as these are both imprinted chromosomes with a parent of origin effect. The risk of heterodisomy is dystrophy. All are due to variable numbers of triplet repeats within the gene. The transmitting parent may influence the chance of expansion. Inheritance patterns: non-Mendelian inheritance
Mitochondrial
All mitochondria are inherited maternally and they have their own genome. If a gene is transcribed from the mitochondria, all children of an affected mother are at risk, but the risk is impossible to predict, as it is likely that both normal and mutated mitochondria will both be present (heteroplasmy). Many genes that are coded for by the nuclear genome are transcribed in the mitochondria and in this case would follow Mendelian inheritance patterns.
Parent of origin affect
Uniparental disomy
Deletion from one parent having a different effect depending if of maternal or paternal origin.
Epigenetic
All cells in an individual with an altered gene expression pattern that does not affect the DNA structure and therefore will not be passed on to future generations. Known genetic disorder within the family
Confirm the diagnosis in the affected patient within the family
Letter of confirmation needed or follow-up from clinician involved in affected patient’s care.
What is the risk to the present pregnancy?
• Does this risk justify prenatal diagnosis?
• Would a termination of pregnancy be considered by the couple for this disease?
• Would a diagnosis alter the management of the pregnancy, labour or early neonatal care?
Type of prenatal diagnosis to be considered
• Invasive prenatal diagnosis
• Molecular: confirmation of parental carrier status and affected patient’s mutations essential. Check that the laboratory will undertake prenatal diagnosis and if they need extra samples. Most laboratories will require parental samples to check for maternal contamination. Some tests take longer than others and therefore prompt testing is advisable for example myotonic dystrophy and fragile X syndromes.
• Enzyme diagnosis: confirmation of enzyme diagnosis in affected patient, parental enzyme levels may be necessary as in some diseases such as metochromatic leukodystrophy there maybe abnormally low levels of the enzyme even in carriers due to rapid metabolism of the enzyme.
• Cytogenetic: karyotype report in affected/carrier individuals to look at chromosome breakpoints, these maybe more difficult to visualise in prenatal samples and therefore may require FISH or molecular cytogenetic techniques for accurate analysis. (see further under chromosome abnormalities)
• Non-invasive prenatal diagnosis
• Ultrasound: what gestation is it possible to identify the abnormalities?
The variability of the syndrome features needs to be considered.
— Certain conditions can only be visualized later as there are no phenotypic abnormalities early on such as achondroplasia
— Others cannot be seen due to the resolution of the ultrasound and evolving development of the normal structure such as the corpus callosum (see later for US diagnosis)
• MRI scan: specialized investigation that normally aids rather than replaces ultrasound as the investigation of choice.
• Non-invasive prenatal diagnosis (NIPD) by free fetal DNA (ffDNA) (see below).
Non-invasive prenatal diagnosis
ffDNA
Three per cent of free DNA in maternal blood is of fetal origin and these short segments of DNA (100–200 kb) are mainly in the form of nucleosomes. This free DNA needs to be enriched for the fetal component to be identified from the maternal ffDNA.
At the present time we can use this for fetal sexing (e.g. fetus at risk of an X-linked disorder) to avoid invasive prenatal diagnosis (PND) and for sexing in congenital adrenal hyperplasia for steroid treatment of possible affected females only. It is also used for Rhesus genotyping of the fetus in Rhesus-negative mothers.
Other uses of this technology at the time of going to press are still in the research setting. New dominant mutations suspected on ultrasound can be identified if there is a single mutation causing the disease, such as in achondroplasia and Apert’s syndrome. Paternally inherited mutations can also be looked for in maternal blood. If a couple are at risk of having a child with an autosomal recessive disease, then providing the parents carry different mutations, i.e. an affected baby would be a compound heterozygote, it would be possible to look for the paternal allele in the maternal blood.
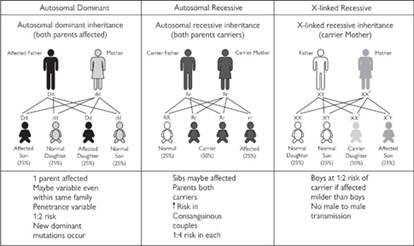
Fig. 7.7.1 Mendelian inheritance patterns. See also colour plate section.
At the present time much work is going on to develop a test for chromosomal aneuploidy by ffDNA. Such tests may not be able to detect all trisomic fetuses, as they require heterozygosity of the markers used. Genetic abnormality suggested from screening investigations
Increased nuchal translucency (NT)
• 3 mm should automatically generate a full karyotype.
• >3.5 mm: a 20-week detailed cardiac scan and detailed anomaly scan by a fetal medicine specialist should be performed.
• The higher the NT the poorer the prognosis, whatever the underlying cause; congenital heart disease and diaphragmatic hernias are the most consistent associations but many rare genetic syndromes have been associated with increased NT.
Abnormal first trimester serum biochemistry
• Very low pregnancy-associated plasma protein-A (PAPP-A): pregnancy at increased risk of placentation problems
• ↓oestriol steroid sulphatase deficiency (X-linked ichthyosis), may have microdeletion at Xp22.3 therefore need to check chromosomes and FISH analysis for this.
• ↓PAPP-A in second trimester maybe associated with Cornelia de Lange syndrome.
Second trimester ultrasound abnormalities (at the 20-week scan)
Confirmation of abnormality is strongly recommended in a fetal medicine unit.
Head
• Microcephaly: check parental head circumference, examine brain structure, follow growth, as frequently it is progressive. Fetal MRI in the third trimester to check for neuronal migration abnormalities, exclude congenital infection, and examine rest of the baby. Many syndromes— including chromosomal and syndromes such as Cornelia de Lange syndrome, frequently do not present until late in gestation and many are of postnatal onset. Familial recurrence is easily missed. Boys head circumference is normally bigger than girls even antenatally
• Macrocephaly: check parental head circumference, examine brain structure, if normal look at overall growth of baby. Generalized macrosomia in second trimester think of Simpson Golabi Behmel Syndrome; late macro-somia consider other overgrowth syndromes—Beck-with Weidemann, Soto’s, isolated macrocephaly – PTEN mutations, Gorlin’s syndrome:
Simpson Golabi Behmel Syndrome X-linked recessive
• Prenatal: early-onset overgrowth, congenital heart disease (VSD/other), diaphragmatic hernia, cleft lip and palate.
• Postnatal: mental retardation, supernumerary nipples, cardiac dysrythmias, tumour risk, e.g. hepatoblastoma, sarcoma.
Beckwith Wiedemann
Complicated genetics, mostly sporadic secondary to imprinting defects. Autosomal dominant and chromosomal abnormalities around 11p15 account for a proportion.
• Prenatal: small omphalocele, macroglossia, nephromegaly, overgrowth
• Postnatal: hypoglycaemia. The majority have normal IQ, tumour risk, Wilms and hepatoblastoma depending on genetic cause of Beckwith Weidemann syndrome. Offer full karyotyping and methylation studies of chromosome 11p15.
Soto’s syndrome
Majority new dominant secondary to NSD mutations on chromosome 5q35
Unlikely to pick up prenatally as no major congenital abnormalities.
• Postnatal: mental retardation, triangular shaped head.
PTEN and Gorlin’s
Both have large heads in the absence of other evidence of overgrowth. PTEN very variable presentations; can be associated with mental retardation and autism and tumour risk. Gorlin’s syndrome is secondary to mutations in the PTCH gene. Shows fusion of ribs occasionally, cleft lip and palate mandibular cysts and propensity to develop basal cell carcinomas.
Abnormal skull shape: consider craniosynostosis syndromes
Apert’s syndrome
The majority are due to new dominant mutations. All are caused by two mutations in FGFR2
• Prenatal: all sutures but lamboidal fused at birth but unsure at what gestation this is apparent. Prominent eyes/proptosis, flat face, cutaneous/bony syndactyly of hands and feet. 3D ultrasound may be helpful. It can be confirmed by fetal DNA studies.
• Postnatal associated with mild to moderate mental retardation.
Pfeiffer syndrome
Autosomal dominant, prenatal lethal form FGFR1 + FGFR2 mutations
• Prenatal: coronal craniosynostosis, Lethal form clover leaf skull, severe proptosis, broad halluces and thumbs, partial hands and feet syndactyly
Saethre Chotzen
Autosomal dominant very variable penetrance
• Prenatal: unlikely to identify any features in absence of family history and even then very unlikely without molecular confirmation of TWIST mutation
• Postnatal: asymmetrical coronal synostosis, ptosis, mild mental retardation, prominent ear crus, skin syndactyly.
Clover leave
Skull may be present with thanatophoric dysplasia (FGFR3 mutation). Look at long bones: telephone handle femur
• Prominent forehead maybe present in achondroplasia.
Brain abnormalities
• Ventriculomegaly: If severe, consider hydrocephalus in a male, look for aqueduct stenosis (X-Linked hydrocephalus) with severe mental retardation. Store DNA for LICAM mutational analysis
• Risk of mental handicap with isolated ventriculomegaly in absence of any other brain abnormality.
• Mild 12–13 mm at 22 weeks 790% normal development
• Moderate 14–15 mm at 22 weeks 760% normal development
• Severe >15 mm at 22 weeks; very high risk of moderate to severe mental retardation
• Agenesis of the corpus callosum. If isolated, risk of mental handicap is around 50%. In the presence of associated abnormalities, it is likely to be nearly 100%
• Cerebellum
• Agenesis of the cerebellar vermis: almost 100% risk of mental handicap. If partial risk is about 25%. Consider Joubert’s syndrome
• Cerebellar hypoplasia: bilateral is associated with poor prognosis, unilateral maybe normal if brain is otherwise normal. Chromosome abnormalities are common.
MRI is useful to look for evidence of lissencephaly in the third and LIS1 + DCX1 lissencephaly syndromes + Walker Warburg Syndrome.
• Other syndromes associated with cerebellar hypoplasia: Smith Lemli Opitz syndrome. Prenatal: ambiguous genitalia in male, polydactyly; Postnatal: mental retardation, two or three toe syndactyly, microcephaly, ptosis. If suspected, maternal urine can be tested for seven dehydrosteroids.
Carbohydrate deficient glycoprotein syndrome (CDG): Many different types have been described. This is unlikely to be picked up prenatally in the absence of family history. Cannot screen antenatally using isoelectric focusing of transferring. Phosphomannomutase levels for CDG1 can be measured
• Encephalocele
Small encephalocele with no apparent brain tissue good prognosis
Other encephaloceles high risk of mental handicap
• + polycystic kidneys and polydactyly: Meckel Gruber syndrome
• + hydrocephalus and lissencephaly: Walker Warburg Syndrome.
Short Long bones
(see Chapter 7.12 on skeletal dysplasia)
Other skeletal problems
• Talipes
• Diastrophic dysplasia
• Relative macrocephaly
• Flat face
• Chondrodysplasia punctata (look for punctuate calcification particularly in knee + ankle + spine)
• Polydactlyly
• First trimester short rib polydactyly syndromes, from 15 weeks Ellis Van Creveld look for congenital eart disease), Jeunes asphyxiating thoracic dystrophy
• Late onset short limbs achondroplasia +frontal bossing.
Limb reduction defects
• Terminal transverse defect single limb good prognosis
• Radial ray defect
• + cardiac abnormalities, Holt Oram syndrome
• Chromosome breakage (need to contact laboratory prior to taking sample to arrange special transfer to appropriate laboratory) Fanconi syndrome,
• + vertebral + renal abnormalities consider VATER syndrome if chromosome breakage is normal.
• Limb reduction with ectrodactyly
• Look for IUGR, low PAPP-A in mid-trimester Cornelia de Lange syndrome.
Arythrogryposis
Talipes normally isolated but need to undertake second scan 4 weeks after first to check isolated and no progression to another joints
If multiple joints:
• Joint dislocations: Larsen’s syndrome, nail patella
• Fetal constraint
• Prelabour rupture of membrane
• Uterine abnormalities (rare)
• Fetal akinesia sequence
• Primary muscle disorder: congenital muscular dystrophy, congenital myopathy, may have polyhydramnios.
• Primary neurological disorder: many disorders including haemorrhage/anoxia. Check for hepatosplenomegaly (neurometabolic).
• Maternal + fetal myotonic dystrophy. This maybe asymptomatic in the mother check for premature cataracts, shake her hand for evidence of myotonia, DNA for myotonin expansion.
• Maternal myasthenia gravis, may be asymptomatic, ask about symptoms muscle weakness especially when tired; measure maternal anti cholinesterase antibodies. If diagnosis confirmed seek specialist advice for Rx.
Polyhydramnios is a poor prognostic sign as suggests baby is unable to swallow and baby may die soon after birth from pulmonary hypoplasia.
Fetal hydrops see Chapter 7.17
• Fetal anaemia (high middle cerebral artery Doppler)
• Rhesus disease + other blood group incompatibility (haemolysis)
• Parvovirus (aplastic anaemia)
• Haemorrhage
• Blackfan diamond syndrome (aplastic anaemia).
• Cardiac causes
• Cardiac conduction defects
• Long QT (maternal + paternal ECGs)
• Anti-Rho antibodies (maternal systemic lupus erythematosis)
• Cardiomyopathy
• Noonan’s syndrome (look for ↓ NT, pulmonary valve disease) PTPN11 mutation
• Costello syndrome (look for ↓NT, early onset polyhydramnios) HRas mutation
• Neurometabolic, look for hepatosplenomegaly Gaucher’s disease, Nieman pick type C, carbohydrate-deficient glycoprotein syndrome
• Obstructive
• Idiopathic.
Fetal renal disease
Bilateral renal agenesis
Universally lethal, associated with oligohydramnios. 25% will be secondary to a de novo mutation in RET. Mainly sporadic but need to undertake. Parental renal ultrasound as may have renal abnormality which will affect recurrence risk, parents will need to be examined for evidence of branchio Oto renal syndrome (branchial clefts, ear abnormalities).
Unilateral renal agenesis
Good prognosis, look for single umbilical artery, features of VACTERL (vertebral, cardiac, tracheo-oesphageal fistula, renal defects, limb defects (radial ray) will not see anal abnormalities.
Polycystic kidneys
Bright kidneys on ultrasound if large and oligohydramnios likely diagnosis autosomal recessive polycystic kidney disease (ARPKD) but differential includes multiple acyl-CoA dehydrogenase deficiency (MADD). PM essential to differentiate different diseases.
Bright kidneys with normal amniotic fluid which may be large need to consider autosomal dominant polycystic kidney (ADPKD) look at parental kidneys, prognosis is good, renal function tends to be normal in childhood but follow up required with paediatric nephrologists for prompt management of hypertension and UTIs.
Bright kidneys with development of second trimester polyhydramnios, check maternal α-fetoprotein if ↓ consider Finnish nephropathy (if parents of Finnish ancestry can undertake mutational analysis to confirm otherwise not possible). Gastrointestinal abnormalities
Exomphalos
Thirty per cent associated with chromosome abnormalities, small exomphalos and those with other abnormalities highest risk. Need to consider Beckwith Weidemann syndrome 11p15 methylation studies can be undertaken to look for this.
Gastroschisis
Normally isolated in young mothers, incidence increasing for unknown reasons often associated with IUGR, may require early delivery. Associations:
• Hyperechogenic bowel
• Cystic fibrosis
• Trisomy 21
• Fetal infection.
Ambiguous genitalia
Discordance between fetal karyotype and ultrasound appearance, occasionally identified on ultrasound in absence of karyotypic known sex.
Check with laboratory that no error in report writing has taken place or sampling; take maternal and paternal blood to check for maternity/paternity. FISH studies for SRY gene. If necessary undertake ffDNA studies on mother.
46, XX with male genitalia
• Congenital adrenal hyperplasia
• SRY translocated to X chromosome.
46XY with female genitalia
• Campomelic dysplasia: look at long bones
• Smith Lemli Optiz syndrome
• Androgen insensitivity syndrome (AIS) (may have family history).
Further investigations during the pregnancy
• Fetal blood sample
• FBC for anaemia and platelets, can identify immunodeficiency syndromes but first trimester diagnosis by DNA diagnosis should be used if available
• Viral infections PCR for virus and immunoglobulins
• Chromosomes
• Amniocentesis
• Chromosomes if not performed
• Amniotic fluid for culture of fibroblasts.
Non-invasive
• 3D ultrasound, can see facial features more clearly may help to define a syndrome more clearly
• Fetal MRI
• Brain abnormalities more clearly identified, most useful in third trimester, brain pathology difficult at post mortem and therefore even if termination is going to be performed it maybe useful to clarify abnormalities:
• Lissencephaly
• Neuronal migration defects (nodular heterotopias, polymicrogyria)
• Tubers in tuberculus sclerosis
• Maternal blood sample
• Viral titres: compare with booking blood samples (may not show evidence of infection). If strongly suspicious, may need invasive test
• Maternal antibodies
• ABO/Rh, platelets—fetal anaemia, ventriculomegaly
• Anti-Rho: fetal Heart block
• anticholinesterase antibodies: arythrogryposis.
Further investigations on the baby
Feticide
If a feticide is being performed and if DNA might be needed, store a sample at this stage. If a karyotype has not been undertaken, take blood. Amniotic fluid may need to be used for metabolic investigations, store at–20°C.
Delivery
Live birth
Cord blood can be used for many investigations. Planning ahead is essential with correct bottles and laboratory addresses. Discussion with neonatal unit on possible complications and interventions that can be anticipated.
After termination of pregnancy
Post mortem
Post mortem is essential to try and clarify underlying diagnosis, it may not clarify this but without a certain prenatal diagnosis recurrence risks will definitely be inaccurate. For example many syndromes cause polycystic kidneys and a ductal plate malformation needed to confirm ARPKD from ADPKD and other cystic kidney diseases. Skeletal Dysplasia need X-rays and histology for clarification of the type.
Placental histology may be useful in clarifying evidence of placental insufficiency secondary to pre-eclampsia, chronic intervillositis, or chromosome mosaicism. Further reading
Bakalis S, Sairam S, Homfray T, et al. Outcome of antenatally diagnosed talipes equinovarus in an unselected obstetric population. Ultrasound Obstet Gynecol 2002;20:226–9.
Boltshauser E. Cerebellum-small brain but large confusion: a review of selected cerebellar malformations and disruptions Am J Med Genet A 2004;126A:376–85.
Brady AF, Pandya PP, Yuksel B, et al. Outcome of chromosomally normal livebirths with increased fetal nuchal translucency at 10–14 weeks’ gestation. J Med Genet. 1998;35:222–4.
Estroff JA, Scott MR, Benacerraf BR. Dandy-Walker variant: prenatal sonographic features and clinical outcome. Radiology 1992;185:755–8.
Fratelli N, Papageorghiou AT, Prefumo F, et al. Outcome of prenatally diagnosed agenesis of the corpus callosum. Prenat Diagn 2007;27:512–7.
Has R, Ermisş H, Yüksel A, et al. Dandy-Walker malformation: a review of 78 cases diagnosed by prenatal sonography Fetal Diagn Ther 2004;19:342–7.
Joo JG, Toth Z, Beke A, et al. Etiology, prenatal diagnostics and outcome of ventriculomegaly in 230 cases Fetal Diagn Ther 2008;24:254–63.
Mehta TS, Levine D. Imaging of fetal cerebral ventriculomegaly: a guide to management and outcome. Semin Fetal Neonatal Med 2005;10:421–8.
Niesen CE. Malformations of the posterior fossa: current perspectives Semin Pediatr Neurol. 2002;9:320–34.
Pandya PP, Kondylios A, Hilbert L, et al. Chromosomal defects and outcome in 1015 fetuses with increased nuchal translucency. Ultrasound Obstet Gynecol 1995;5:15–9.
Papageorghiou AT, Fratelli N, Leslie K, et al. Outcome of fetuses with antenatally diagnosed short femur. Ultrasound Obstet Gynecol 2008;31:507–11.
Poretti A, Wolf NI, Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood. Eur J Paediatr Neurol 2008;12:155–67. Poretti A, Prayer D, Boltshauser E. Morphological spectrum of prenatal cerebellar disruptions. Eur J Paediatr Neurol 2008 Oct 20. [Epub ahead of print]
Scott RH, Douglas J, Baskcomb L, et al. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with over-growth and growth retardation. J Med Genet. 2008;45:106–13.
Shackleton CH, Marcos J, Palomaki GE, et al. Dehydrosteroid measurements in maternal urine or serum for the prenatal diagnosis of Smith-Lemli-Opitz syndrome (SLOS). Am J Med Genet A 2007;143A:2129–36.
Vissers LE, de Vries BB, Osoegawa K, et al. Array-based comparative genomic hybridization for the genomewide detection of sub-microscopic chromosomal abnormalities. Am J Hum Genet 2003;73:1261–70.
Volpe P, Paladini D, Resta M, et al. Characteristics, associations and outcome of partial agenesis of the corpus callosum in the fetus. Ultrasound Obstet Gynecol 2006;27:509–16.
Wolstenholme J. Confined placental mosaicism for trisomies 2, 3, 7, 8, 9, 16, and 22: their incidence, likely origins, and mechanisms for cell lineage compartmentalization. Prenat Diagn 1996;16:511–24.
Wright CF, Burton H. The use of cell-free fetal nucleic acids in maternal blood for non-invasive prenatal diagnosis. Hum Reprod Update 2008. Internet resources
Gene Reviews: www.geneclinics.org
OMIM: www.ncbi.nlm.nih.gov
BSHG: www.bshg.org.uk
National Metabolic Biochemical Network: www.metbio.net
Fetal abnormalities: face
Facial clefts
Definition
Facial clefts encompass a wide spectrum of defects (unilateral, bilateral and less commonly midline) usually involving the upper lip, the palate, or both. The typical cleft lip will appear as a linear defect extending from one side of the lip into the nostril. Cleft palate associated with cleft lip may extend through the alveolar ridge and hard palate, reaching the floor of the nasal cavity or even the floor of the orbit. Isolated cleft palate may include defects of the hard palate, the soft palate, or both. Cleft lip and palate is unilateral in about 75% of cases and the left side is more often involved than the right side. Cleft palate without cleft lip is a distinct disorder.
Prevalence
Facial clefting is found in about 1 per 800 births. In about 50% of cases both the lip and palate are defective, in 25% only the lip and in 25% only the palate is involved.
Aetiology
Cleft lip with or without cleft palate is usually an isolated condition, but in 20% of cases it is associated with one of many genetic syndromes. Isolated cleft palate is a different condition. All forms of inheritance have been described, including autosomal dominant, autosomal recessive, X-linked dominant and X-linked recessive. Associated anomalies are found in about 50% of patients with isolated cleft palate and in about 15% of those with cleft lip and palate. Chromosomal abnormalities (mainly trisomy 13 and 18) are found in 1–2% of cases and exposure to teratogens (such as antiepileptic drugs) in about 5% of cases. Recurrences are type specific; if the index case has cleft lip and palate there is no increased risk for isolated cleft palate, and vice versa. Median cleft lip, which accounts for about 0.5% of all cases of cleft lip, is usually associated with holoprosencephaly or the oral-facial-digital syndrome.
Clinical approach: diagnosis
The sonographic diagnosis of cleft lip depends on demonstration of a groove extending from one of the nostrils inside the lip and possibly the alveolar ridge and anterior palate. Both axial and coronal planes can be used (Fig. 7.8.1). Three-dimensional ultrasound may be useful (Fig. 7.8.2). The diagnosis of isolated cleft palate is difficult.
Prognosis
Minimal defects, such as linear indentations of the lips or submucosal cleft of the soft palate, may not require surgical correction. Larger defects cause cosmetic, swallowing, and respiratory problems. Recent advances in surgical technique have produced good cosmetic and functional results. However, prognosis depends primarily on the presence and type of associated anomalies. Micrognathia
Definition
Mandibular hypoplasia causing a receding chin. Differentiation from cases in which the mandible is of normal size but is dislocated posteriorly (retrognathia) may be difficult.
Prevalence
Micrognathia is found in about 1 per 1000 births.
Aetiology
Micrognathia is usually associated with genetic syndromes (such as Treacher Collins, Robin and Robert syndromes), chromosomal abnormalities (mainly trisomy 18 and triploidy), and teratogenic drugs (such as methotrexate). The Robin anomalad (severe micrognathia, glossoptosis and a posterior cleft palate or an arched palate) may be a sporadic isolated finding (in about 40% of cases) or it may be associated with other anomalies or with recognized genetic and non-genetic syndromes.
Clinical approach: diagnosis
Several approaches to diagnosis have been suggested, including measurement of the mandible. However, most would use a subjective evaluation of the profile in which there is evidence of a receding chin (Fig. 7.8.3). In severe cases, glossoptosis can also be noted and increases the likelihood of the condition.
Prognosis
This depends on the presence of associated anomalies. Severe micrognathia can be a neonatal emergency due to airway obstruction by the tongue in the small oral cavity.

Fig. 7.8.1 Axial planes of the maxilla in fetuses with facial clefts: (a) isolated cleft lip: the alveolar ridge is intact albeit irregular in shape as frequently happens in these cases; b) unilateral cleft lip and palate: the defect only extends to the alveolar ridge; note that one toothbud is missing but that the secondary palate does look intact; these defect is frequently referred to as cleft alveolus; (c) unilateral cleft lip and palate: the defect is seen extending to the secondary palate: (d) bilateral cleft lip and palate; the anterior protrusion of the central portion of the maxilla (or premaxilla) indicates that the defect expends posteriorly to the secondary palate.
Fig. 7.8.2 Three dimensional ultrasound of cleft lip in rendering of the surface (a, b, c) and skeletal mode (d, e, f); (a, c) unilateral cleft lip; b, e) unilateral cleft lip and palate; c, f) bilateral cleft lip and palate.

Fig. 7.8.3 Severe micrognathia in bidimensional and three-dimensional ultrasound.
If prenatal diagnosis is made, a paediatrician should be present in the delivery room and be prepared to intubate the infant. Further reading
Berge SJ, Plath H, Van de Vondel PT, et al. Fetal cleft lip and palate: sonographic diagnosis, chromosomal abnormalities, associated anomalies and postnatal outcome in 70 fetuses. Ultrasound Obstet Gynecol 2001;18:422–31.
Bromley B, Benacerraf BR. Fetal micrognathia: associated anomalies and outcome. J Ultrasound Med. 1994 Jul;13(7):529–33.
Ghi T, Perolo A, Banzi C, et al. Two-dimensional ultrasound is accurate in the diagnosis of fetal craniofacial malformation. Ultrasound Obstet Gynecol 2002;19:543–51.
Rotten D, Levaillant JM, Martinez H, et al. The fetal mandible: a 2D and 3D sonographic approach to the diagnosis of retrognathia and micrognathia. Ultrasound Obstet Gynecol. 2002;19:122–30.
Rotten D, Levaillant JM. Two- and three-dimensional sonographic assessment of the fetal face. 2. Analysis of cleft lip, alveolus and palate. Ultrasound Obstet Gynecol 2004;24:402–11.
Fetal abnormalities: gastrointestinal system Anterior abdominal wall defects
Definition
Defect in the anterior abdominal wall allowing herniation of intra-abdominal contents.
Epidemiology
Exomphalos (also referred to as omphalocele) prevalence of 1:4000 births; gastroschisis prevalence of 1:3000 births.
Body stalk anomaly (1:10 000 births with some studies reporting 1:14 000–42 000). Exstrophy and epispadias complex: bladder exstrophy (1 in 30 000 births) and cloacal exstrophy (which can be part of omphalocele-exstrophy-imperforate anus-spinal defects (OEIS) complex) (1 in 200 000 births) are rarer conditions.
The prevalence of gastroschisis has risen sharply in the last 20 years and is strongly associated with young maternal age. Mothers under 20 years old have a 12-fold increase in risk. Exomphalos in contrast increases with advanced maternal age.
Pathology
Exomphalos: This is a midline defect of the abdominal closure that involves the cord insertion. There is herniation of small bowel and/or liver, which are both wrapped in a two-layered sac formed by the amnion and the peritoneum. Exomphalos can also be a large defect and cases containing stomach, large bowel, bladder, and spleen have been reported. Rupture of the sac can occur and in such cases differential diagnosis with gastroschisis can be difficult. Less often there is associated failure of the cephalic or caudal embryonic folds with Pentalogy of Cantrell and bladder/cloacal exstrophy respectively.
Gastroschisis: This involves evisceration of bowel through a small abdominal wall defect, usually to the right of the umbilical cord insertion. The defect is possibly related to abnormal involution of the right umbilical vein.
Body stalk anomaly, or more appropriately, amniotic rupture sequence is characterized by a large body wall defect with fusion of the fetal peritoneum to the amniotic cavity and fetal tethering to the side wall. It is thought to occur as a result of early rupture of the amnion before the coelomic cavity is obliterated.
Bladder exstrophy results from a defect in the caudal fold of the anterior abdominal wall. These range in size from an epispadias to exposure of the posterior bladder wall.
Cloacal exstrophy is usually present as part of multiple defects present in OEIS complex that comprises exomphalos, bladder exstrophy, imperforate anus, and spinal defects.
Aetiology
Exomphalos: In the majority of cases the aetiology is sporadic. There is a strong association with chromosomal abnormalities, mainly trisomy 18 and 13 (about 50% at 12 weeks, 30% at midgestation and 15% of neonates). The risk may be highest with a small exomphalos; a large exomphalos containing the liver is more rarely associated with chromosomal abnormalities. Associated structural abnormalities are present in 60–80% of cases and in 40% of cases with normal karyotype (mainly cardiac, other gastrointestinal (GI) and urogenital malformations). The association with non chromosomal syndrome is also high and Beckwith–Wiedemann syndrome (suspect if there is fetal macrosomia) is the cause in 15–20% of cases.
Gastroschisis: usually sporadic and isolated, with no association with chromosomal abnormalities. Although in 10–30% of cases other GI tract abnormalities are found, these are mainly bowel atresias due to in utero strangulation and infarction. A link to an environmental teratogens and drug abuse use has been postulated but not proven.
Clinical approach
Ultrasound findings: key points
• The stomach can be visible in the left upper quadrant from 9 weeks’ gestation.
• The integrity of the anterior abdominal wall should be confirmed by demonstrating normal cord insertion.
• Exstrophy can be ruled out by confirming the presence of the fetal bladder in the pelvis.
• Physiological herniation of the midgut into the umbilical sac can be present until 11 weeks’ gestation and any abnormality after this should prompt careful assessment.
• Exomphalos: central, midline sac containing bowel/liver at the level of the umbilicus with the umbilical vessels traversing through the sac (Fig. 7.9.1).
• Gastroschisis: free-floating loops of bowel are seen herniating (cauliflower appearance) lateral to a normal cord insertion (Fig. 7.9.2).
• Body stalk anomaly/amniotic rupture sequence: large midline abdominal defect with severe kyphoscoliosis and a short or absent umbilical cord.
• Bladder/cloacal exstrophy: infraumbilical defect, usually large, a low protruding mass with an absent bladder and normal amniotic fluid volume. In cloacal exstrophy commonly there are lumbar sacral, lower limb and renal anomalies.
Diagnosis key points
The vast majority of anterior abdominal wall defects are now diagnosed in the first or second trimester by routine ultrasound screening (USS). Sensitivity for USS detection is ˜95% (RCOG). Gastroschisis and exstrophy may still present with markedly raised AFP levels.
Investigation
The diagnosis should be confirmed by a Fetal Medicine Specialist.
Invasive testing should be considered, except in the case of isolated gastroschisis where the risk of aneuploidy is low.
Antenatal and delivery
As in all cases of fetal anomaly, multidisciplinary input including the fetal medicine team, geneticists, neonatologists, and surgeons is essential. In particular, early involvement of the paediatric surgical team is particularly important in cases of GI tract abnormalities.
Both exomphalos and gastroschisis are associated with a high risk of fetal growth restriction. Difficulty in accurately measuring abdominal circumference can make monitoring fetal growth more difficult. Therefore, regular growth scans and Doppler/biophysical assessment of fetal wellbeing should take place.
Progressive bowel dilatation has been associated with a poorer prognosis.
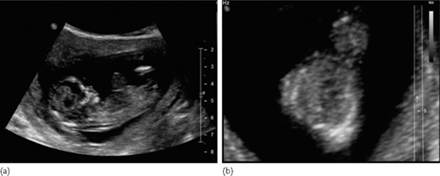
Fig. 7.9.1 (a) Sagittal and (b) axial view of a fetus with exomphalos at 12 weeks.
In particular, the occurrence of bowel obstruction has been recently demonstrated to be correlated with prenatal detection of intra-abdominal bowel dilatation.
Planned delivery in a unit with adequate neonatal care and surgical facilities is advised and fetal growth restriction often mandates induction previous to term.
The majority of cases with GI abnormality are suitable for vaginal delivery. There is no evidence of benefit of delivery by Caesarean section in gastroschisis or exomphalos unless there are other fetal or maternal indications.
Postnatal
Early management involves stabilization of the neonate and immediate covering of the affected area by clingfilm. There may be respiratory compromise with a large exomphalos due to a small chest and pulmonary hypoplasia.
Surgical management involving primary or staged closure of the wall defect can then take place in due course. The intra-abdominal contents may also be slowly reduced over time with a bag and the aid of gravity in a large number of cases.
Prognosis
Exomphalos with normal chromosomes has a survival at 1 year of over 95%.
Gastroschisis has a 90% survival at 1 year with most of the mortality relating to preterm delivery and short gut syndrome in cases with extensive bowel atresia.
Amniotic rupture sequence is almost invariably lethal due to the multiple and severe fetal anomalies.
In bladder and cloacal exstrophy, the prognosis is dependent on the site and size of the defect. Survival has been reported to be over 80% at 1 year, but a variable degree of long-term morbidity is often present. Extensive reconstructive surgery and permanent urinary diversion may be required. Male fetuses may have severely abnormal external genitalia and almost in all cases despite size, there is a certain grade of urinary continence and reproductive problems.
Termination of pregnancy
In the case of an anterior abdominal wall defect with a predictable poor prognosis a termination may be offered. The presence of a severe associated chromosomal abnormality (such as trisomy 18 or 13) may influence this decision.

Fig 7.9.2 (a) sagittal and (b) axial view of the fetal abdomen at 21 weeks showing gastroschisis at the level of the umbilical cord insertion.
Owing to earlier diagnosis, usually in the first trimester, more parents may choose this option. Parents should have the opportunity to discuss their case with both a fetal medicine specialist and a paediatric surgeon prior to making this decision.
A detailed post-mortem is valuable in confirming the diagnosis and providing information for future pregnancies, although most of these cases are sporadic.
Future pregnancies
In general, the majority of these anomalies are sporadic with a low recurrence risk.
The recurrence risk for exomphalos is about 1%. When it is part of a genetic syndrome such as Beckwith–Wiedemann, a definition of the particular syndrome and a consultation with the geneticists is required to define possible increased recurrence risk.
Gastroschisis has a low recurrence risk of ~1%.
Bladder exstrophy is usually sporadic with ~1% recurrence, even though rare familial cases have been reported.
Amniotc rupture sequence and cloacal exstrophy (OEIS complex) are very rare, sporadic, and with a low recurrence risk. GI tract abnormalities
Oesophageal atresia
Definition
Atresia of the oesophagus; this is associated with tracheooesophageal fistula (TEF) in about 90% of cases.
Epidemiology
1:3000–3500 births.
Pathology
Failure of the primitive foregut to divide into the trachea and oesophagus in the fourth week of gestation.
Aetiology
Usually sporadic in nature, and a clear aetiology is unknown in the majority of cases. About 20% are associated with chromosomal abnormalities (trisomy 18, 21 and 13). Other major structural defects are seen in 50% of cases, mainly cardiac and other bowel anomalies. TEF may also occur as part of VACTERL/VATER association (vertebral defects–anal atresia–cardiac anomalies–tracheoesophageal fistula–esophageal atresia–renal defects limb defects).
Duodenal atresia
Definition
Lack of normal canalisation of the duodenal lumen leading to partial (web) or complete obstruction (atresia).
Epidemiology 1 in 5000 births.
Pathology
The lumen of the duodenum in early embryonic life is completely obliterated by proliferating epithelium and is normally canalized by the eleventh week. Failure of this process leads to duodenal atresia, which can also be caused by external compression due to an annular pancreas, or peritoneal bands.
Aetiology
Over 30% of cases are associated with trisomy 21. Other structural abnormalities are seen in up to 50% of cases and these are mainly skeletal, cardiac, renal, and other GI abnormalities.
Intestinal obstruction
Definition
Stenosis or atresia involving the distal small bowel in one or more areas of the GI tract. Equally frequent in the jejunum and ileum.
It also includes anorectal atresia or Hirschprung’s disease.
Epidemiology
Intestinal obstruction occurs in about 1 in 2000 births. Many cases are not diagnosed antenatally and present in the first few days of neonatal life with abdominal distension and vomiting. The more proximal the lesion is the more likely it is to present itself antenatally with polyhydramnios and ultrasound detection of multiple fluid-filled bowel loops.
Jejunal and ileal atresia occur in 1 in 3000 births.
Hirschprung’s disease is rare (1 in 5000 births) and more commonly seen in male fetuses.
Pathology
Small bowel atresias probably result from a vascular insult to the developing bowel during rotation at 6–12 weeks of embryonic life. They are frequently associated with volvulus and malrotation. There may be multiple segments involved; absence of large sections of small bowel is known as ‘apple peel atresia’.
Anorectal atresia results from abnormal cloacal division in the ninth week of development.
Other causes of obstruction include meconium ileus, peritoneal bands, volvulus, and agangliosis of the colon (Hirschprung’s disease). In Hirschprung’s there is absence of the neural crest-derived enteric neural ganglion along a variable length of the intestine.
Aetiology
Jejunal and ileal obstructions are usually sporadic and not associated with chromosomal abnormalities; extraintestinal anomalies are uncommon. It is associated with cystic fibrosis (CF) in 10% of cases but up to 90% of cases if meconium ileus is present.
In contrast, anorectal atresias are frequently associated with chromosomal abnormalities (mostly trisomy 18 and 21) and other structural anomalies in 80% of cases. These include other GI, genitourinary, cardiac, and vertebral anomalies (VACTERL association).
Hirschprung’s is associated with chromosomal abnormalities (particularly trisomy 21) in 10% of cases and other congenital anomalies or syndromes in 20% of cases. The remainder of cases are sporadic and isolated.
Echogenic bowel
Definition
Increased echogenicity (brightness) of the fetal bowel on ultrasound, with brightness similar to that of the bone (iliac crests and lumbar spine).
Epidemiology
A common finding in 1–1.8% of pregnancies in the second and third trimester.
Aetiology
In the majority of cases it is a normal variant and occasionally due to fetal ingestion of blood; however, it is also associated with chromosomal anomalies, congenital infection, CF, and intrauterine growth restriction.
Clinical approach
Ultrasound findings key points
• Polyhydramnios due to intestinal obstruction tends not to present until 25 weeks’ gestation.
• Suspect esophageal atresia in a late second and third trimester fetus with a small or absent stomach and polyhydramnios (Fig.7.9.3).
• Diagnosis can be difficult as the stomach may fill normally with an associated TEF.
• Duodenal atresia has a classic ‘double bubble’ appearance due to dilatation of both the stomach and proximal duodenum (Fig. 7.9.4).
• This is commonly not diagnosed until after 25 weeks, although can sometimes be seen as early as 20 weeks. Continuity of the duodenum with the stomach must be demonstrated to differentiate it from other cystic masses e.g. choledochal cyst.
• Look at the fetal heart; duodenal atresia carries a very high risk of trisomy 21 and may be associated with an atrioventricular septal defect (AVSD) or other markers for trisomy 21.
• Suspect more distal obstruction (jejunal or ileal) with hyperperistalsis in multiple loops of dilated bowel.
• It is not possible antenatally to determine the exact site and cause of the obstruction.
• Additional features such as echogenic bowel or ascites indicate the possibility of meconium peritonitis, which is more common in CF.
• With echogenic bowel assess fetal growth and uterine artery Doppler as this may be the first manifestation of subsequent fetal growth restriction.
History
• Ask about family history of genetic disorders, GI anomalies and in particular CF.
• Prior results of screening or diagnostic tests for aneuploidy should be reviewed.
• With echogenic bowel, ask about any bleeding in pregnancy or invasive testing, maternal illness or rash suggestive of congenital infection, and again family history of CF.
• If polyhydramnios present enquire about shortness of breath and maternal discomfort. Remember to ask about signs and symptoms of threatened preterm labour.

Fig 7.9.3 Axial view of the fetal abdomen showing a collapsed or small stomach.

Fig 7.9.4 The appearance of a double bubble due to proximal small bowel atresia. Note the presence of polyhydramnios.
Investigation
• The diagnosis should be confirmed by a fetal medicine specialist with a careful search for other associated abnormalities.
• Invasive testing for karyotype and CF DNA studies should be offered depending on the findings.
• In cases of echogenic bowel send maternal serum for a congenital infection screen (CMV and toxoplasmosis), perform uterine artery and fetal Doppler studies (increased risk of IUGR) and consider parental CF carrier testing.
Management Antenatal and delivery
• As in all cases of fetal anomaly multidisciplinary input including the fetal medicine team, neonatologists, and geneticists is essential. Early involvement of the paediatric surgical team is particularly important in these cases.
• Polyhydramnios increases the risk of preterm labour and abnormal presentation. Serial amnio reduction may be discussed and performed after a certain gestational age to reduce the risk of preterm labour and maternal discomfort.
• Systemic corticosteroids should be given to promote fetal lung maturity.
• Planned delivery in a unit with adequate neonatal care and surgical facilities is optimal. However, polyhydramnios due to upper GI obstruction will frequently result in unplanned early labour; parents should always be advised to attend the nearest hospital in an emergency.
• The majority of cases with GI abnormality are suitable for vaginal delivery, unless there are other fetal or maternal indications.
Postnatal
Early management involves stabilization of the neonate. There may be respiratory compromise due to severe abdominal distension. In cases of suspected oesophageal fistula and TEF oral feeds should be withheld until after a full assessment by the neonatal team. Surgical management of intestinal obstruction will usually involve resection of the affected bowel segment. The need for a colostomy and subsequent surgery depends on the site and extent of the stenosis or atresia.
Table 7.9.1 Causes of abdominal cysts

Prognosis
The prognosis for GI tract anomalies in the absence of associated chromosomal abnormalities depends largely on the gestation at delivery and the site and extent of any atresia. Large areas of atresia may lead to short gut syndrome, which has a very poor prognosis. Meconium peritonitis is associated with a poor prognosis and over 50% neonatal mortality.
Survival after surgery for isolated duodenal atresia is over 95%. TEF has a variable prognosis due to the high frequency of associated anomalies, but, in isolated cases, survival is excellent. Small bowel atresia has a good final outcome, except for the rare cases of apple peel atresia or multiple atresias. Termination of pregnancy
These cases can be difficult as presentation is often after 24 weeks’ gestation. If there is a very poor prognosis due to chromosomal abnormalities or coexisting defects, termination of pregnancy is an option in some countries, after careful counselling.
Particular ethical dilemmas are raised in cases involving a late diagnosis of trisomy 21. Many centres would not offer late termination in these circumstances.
In any case of pregnancy termination, a detailed postmortem is valuable in confirming the diagnosis and providing information for future pregnancies. Future pregnancies
The majority of cases are sporadic with a low recurrence risk. Rare familial cases of multiple bowel atresias have been reported. Recurrence risk is low for bowel atresia and Hirschprung’s disease (respectively 1–2% and 4%).
Where the affected pregnancy involves CF, and both parents are carriers the recurrence risk is 25% for CF. Abdominal cysts
Abdominal cystic masses are a common ultrasound finding. Renal tract anomalies or bowel dilatation are the most likely cause. Less commonly, cystic structures arise from the biliary tract or from the mesentery or from the ovary in female fetuses.
The origin is usually determined by the position of the cyst and its relation to other organs.
A definitive diagnosis may not be possible antenatally. A summary of causes of abdominal cysts’s is given in Table 7.9.1; renal tract anomalies are not covered here. Further reading
David A, Tan A, Curry J. Gastroschisis: sonographic diagnosis, associations, management and outcome. Prenatal Diagnosis 2008;28;633–44.
Fratelli N, Papageorghiou AT, Bhide A, Sharma A, Okoye B, Thilaganathan B. Outcome of antenatally diagnosed abdominal wall defects. Ultrasound Obstet Gynecol. 2007;30:266–70.
Hyett J, Intra-abdominal masses: prenatal differential diagnosis and management. Prenatal Diagnosis 2008;28;645–55.
Sparey C, Jawaheer G, Barrett AM, Robson SC. Esophageal atresia in the Northern Region Congenital Anomaly Survey, 1985–1997: prenatal diagnosis and outcome. Am J Obstet Gynecol 2000:182:427–31
Twining, McHugo, Pilling. Abdominal and abdominal wall abnormalities In: Textbook of fetal abnormalities, 2nd edn. Edinburgh: Churchill Livingstone Chapter 11.
Wilson RD, Johnson MP. Congenital abdominal wall defects: an update. Fetal Diagn Ther. 2004;19:385–98. Patient resources
GEEPS (Gastroschisis, Exomphalos, Exstrophies Parents Support Group): www.geeps.co.uk
TOFS (Tracheo-Oesophageal Fistula Support): www.tofs.org.uk Fetal abnormalities: limbs Club foot
Definition
Malformation of the ankle joint characterized by a fixed position of the foot held in adduction, supination, and varus positions, in the classical type namely talipes equinovarus.
Incidence
Approximately 1 in a 1000.
Aetiology
May be isolated (commonest form) or complex due to an underlying chromosomal abnormality, neuromuscular and skeletal disorder or genetic syndrome.
Ultrasound features
Usually identified at the 18–23-week scan. In the normal foot, the foot is in a plane that is perpendicular to the long axis of the tibia and fibula. In talipes, the plantar aspect of the fetal foot is imaged in the same longitudinal plane as the tibia and fibula (Fig. 7.10.1).
It is important to do a full fetal survey to assess whether the talipes is a manifestation of a generalized disorder as listed in the box or an isolated condition. Additionally, serial scans need to be organized to assess if there are other joints showing progressive developmental abnormality as in neuromuscular disorders and to assess the liquor volume, as muscular disorders can impair the swallowing of the fetus.
Management
Antenatal: In isolated talipes, especially in the presence of low risk from previous first trimester screening, the pregnancy should be treated as any other low-risk pregnancy. However, in the absence of any screening or in the presence of other structural abnormalities, invasive prenatal diagnosis should be offered to rule out chromosomal associations.
Some conditions that are associated with club foot deformity in the fetus
Chromosome abnormalities
Neuromuscular disorders
Meningomyelocele
Pena shokeir phenotype
Arthrogryposis multiplex cogenita
Skeletal dysplasias
Campomelic dysplasia
Diastrophic dysplasia
Ellis–van Creveld syndrome
Genetic syndromes
Multiple pterygium syndrome
Larsen syndrome
Smith–Lemli–Opitz syndrome

Fig. 7.10.1 Image of a fetal foot with talipes: the foot is in the same plane as the two long bones of the lower limb.
Postnatal: Mild forms of talipes may not require any treatment. More severe forms will require physiotherapy (traditional or Ponseti technique). Surgical correction is now rarely used as results from the Ponseti technique are excellent.
Prognosis
Depends on underlying aetiology. In isolated talipes, prognosis is good although surgical correction may be needed in some cases. Ectrodactyly
Definition
Ectrodactyly, by definition describes a diverse group of hand and foot malformations that could vary from the absence of one digit to absence of all but one digit. It is also known as split hand and foot malformation and in the typical form is characterized by a median cleft of the hands and/or feet and may be associated with syndactyly, and aplasia and/or hypoplasia of the phalanges, metacarpals, and metatarsals.
Incidence
Very rare, about 1:20 000 live births.
Aetiology
It may occur as an isolated feature or as a part of a genetic syndrome (Table 7.10.1), almost all of which have a dominant mode of inheritance.
Table 7.10.1 Clinical features of some ectrodactyly syndromes

Ultrasound features
Typically identified at the time of the 18–23-week scan, although earlier diagnosis is reported. The findings are that of a V- or U-shaped cleft in the middle of the fetal hands and/or legs (Fig. 7.10.2). There is usually a paucity of digits, which may be due to actual absence of the digits or due to syndactyly. There may be additional features as mentioned in Table 7.10.1.
Management
Antenatal: Should include counselling for the parents in the presence of a multidisciplinary team comprising an obstetrician, fetal medicine consultant, genetics consultant and a paediatric plastic surgeon. The session should highlight the likely inheritance and any identifiable syndrome, the role of invasive prenatal diagnosis and the need for surgical correction. Some parents may opt for pregnancy termination based on the features identified.
Postnatal: The anatomical deformity will require surgical correction. For the ectrodactyly, the main aim of the surgery is to establish successful opposition of the two main digits, as complete normality of hand anatomy can usually not be restored. Owing to the possibility of associated renal tract abnormalities, prophylactic antibiotics are often advocated until normality of the renal tract can be established.
Prognosis
In the familial type of ectrodactyly, the outcome tends to be good with normal mentation and development. Morbidity caused by the associated hearing loss and ocular manifestations owing to lacrimal gland involvement in the syndromic patients should be considered. Radial aplasia
Definition
Radial aplasia or hypoplasia is characterized by complete or partial absence of the radius and/or radial ray structures. This may manifest as a single forearm bone in the fetus with or without an absent thumb and abnormal positioning of the fetal hand. These abnormalities are collectively called radial ray defects.
Incidence
Rare: radial ray defects occur in 1 in 30 000 live births.
Aetiology
Radial ray defects and associated anomalies encompass a group of disorders with most defects being unilateral and sporadic while bilateral defects are more likely to be part of a multiple malformation syndrome (Table 7.10.2). It would be useful to identify any medication that the woman has been taking during the course of the pregnancy such as Valproates.
Table 7.10.2 Associated features of some syndromes that may present with radial ray defects

Ultrasound features
Usually identified at the 18–23-week anomaly scan. The findings are either absent or hypoplastic radius, with only bone identified in the fetal forearm (Fig. 7.10.3). This defect may be uni- or bilateral with radial deviation of the hand. There may be associated aplasia or hypoplasia of the thumb. As with any other structural abnormality, a thorough fetal survey is essential in making an assessment and a diagnosis.
Management
Antenatal management should aim for making a diagnosis if possible of the underlying syndromes as outline above. Invasive prenatal diagnosis would be required to clarify the karyotype, chromosomal breakage disorders, and fetal thrombocytopenia. Genetic counselling in these cases would be mandatory.

Fig. 7.10.2 Image showing ectrodactyly in the fetal hand, with a V-shaped cleft in the hand and paucity of digits.

Fig. 7.10.3 Image showing single forearm bone with abnormal positioning of the fetal hand.
Fig. 7.10.4. Image showing normal number of fingers in (a) and postaxial polydactyly in the hand in (b).
Prognosis
Isolated unilateral radial ray defects tend to have a good prognosis as the vast majority of them are due to nonsyndromic limb reduction defects. The syndromic cases have varied outcome. In the presence of other structural malformations, most parents tend to opt for termination. Polydactyly
Definition
Polydactyly is defined as the presence of one or more extra digits in the hands (Fig. 7.10.4) or feet (Fig. 7.10.5). It is classified as pre-axial if the extra digit is on the radial or tibial side and as postaxial if it is on the ulnar or fibular aspects of the hand and foot respectively.
Incidence
Varies between 1:3000 to 10 000 with postaxial being more common.
Aetiology
The vast majority of polydactyly that are identified antenatally are isolated and familial. Polydactyly is thought to be a feature of many genetic syndromes some of which are listed in the Table 7.10.3.
Ultrasound features
Extra digits are noted on hands and/or feet (Fig. 7.10.4). These digits may either be pre- or postaxial and may or may not have bony elements in them. Occasionally, these digits are seen fused with the normal digits presenting as syndactyly.
Management
As with any other abnormality, a thorough search is mandatory to exclude other structural abnormalities. As indicated above in the table, coexisting abnormalities would point towards a genetic syndrome.
If the polydactyly is isolated, the family should be reassured and given information about postnatal surgical management that may be required. Depending on the associated anomaly pattern, invasive prenatal diagnosis will have to be considered.
Prognosis
Polydactyly, in the absence of any other abnormality is associated with an excellent prognosis. If it is not isolated, the underlying aetiology and the associated abnormalities would dictate the prognosis. Amniotic band syndrome
Definition
Amniotic band syndrome refers to a group of congenital anomalies that are thought to be a consequence of amniotic bands that adhere to various organs, more commonly to the fetal extremities, resulting in amputations or peripheral nerve damage due to ischaemia. The amniotic bands can cause extensive disruptions in the craniofacial and truncal regions as well with the fetus presenting with bizarre clefts and abnormalities.

Fig. 7.10.5. Image showing postaxial polydactyly in the foot in (a) and normal number of toes in (b).
Table 7.10.3

Incidence
Incidence varies from 1:1200 to 15000.
Aetiology
The exact underlying aetiology still remains unknown, although some authors believe that this syndrome is caused by early amnion rupture with the fetal parts being exposed to the sticky chorionic membrane. As a result, fibrous band-like adhesions are thought to be formed between the chorion and the exposed fetal parts, usually the extremities. These bands then wrap around the extremity causing ischemic changes in the distal parts, or causing transverse amputations across the limbs or digits.
Ultrasound features
The presenting feature is variable and may be an oedematous arm distal to a constriction, amputated appearance of the extremities (Fig. 7.10.6) or digits, or abnormal posturing of the hands or feet that are oedematous. These may be associated with bizarre malformations of the fetal trunk, head, and neck. Amniotic bands may be noted at the line of constriction in the extremity with or without Doppler evidence of ischaemia distal to the occlusion.

Fig. 7.10.6 Image of the fetal upper limb showing transverse amputation at the forearm.
Management
The presence of abnormalities as described should prompt the diagnosis of amniotic band syndrome. It is important to ascertain the extent of damage with particular reference to level of amputation and degree of ischemia in the affected limb. It may be possible to limit the damage by fetoscopic surgical release of the constricting membrane in rare cases. There are also anecdotal reports of spontaneous release of the constricting membrane with complete resolution of the changes in the limb.
Prognosis
Isolated limb abnormalities are usually non-lethal and compatible with normal life. Some abnormalities are likely to cause physical deformity and limitation of function. This may not be acceptable to some parents and they may opt for termination of the pregnancy. The prognosis depends on the severity of the abnormality and the possibility of reconstruction to near normal state with plastic surgery. Multiple malformations due to amniotic bands may be lethal or incompatible with normal life. Further reading
Bakalis S, Sairam S, Homfray T, et al. Outcome of antenatally diagnosed talipes equinovarus in an unselected obstetric population. Ultrasound Obstet Gynecol 2002;20:226–9.
Bromley B, Shipp TD, Benacerraf B. Isolated polydactyly: prenatal diagnosis and perinatal outcome. Prenat Diagn 2000;20:905–8.
Canto MJ, Cano S, Palau J, Ojeda F. Prenatal diagnosis of clubfoot in low-risk population: associated anomalies and long-term outcome. Prenat Diagn 2008;28:343–6.
Cox H, Viljoen D, Versfeld G, Beighton P. Radial ray defects and associated anomalies. Clin Genet 1989;35:322–30.
Elliott AM, Evans JA, Chudley AE. Split hand/foot malformation (SHFM). Clin Genet 2005:68:501–5.
Kennelly MM, Moran P. A clinical algorithm of prenatal diagnosis of Radial Ray Defects with two and three dimensional ultrasound. Prenat Diagn 2007;27:730–7.
Keswani SG, Johnson MP, Adzick NS, et al. In utero limb salvage: fetoscopic release of amniotic bands for threatened limb amputation. J Pediatr Surg 2003;38:848–51.
Leung KY, MacLachlan NA, Sepulveda W. Prenatal diagnosis of ectrodactyly: the ‘lobster claw’ anomaly. Ultrasound Obstet Gynecol 1995;6:443–6.
Paladini D, Foglia S, Sglavo G, Martinelli P. Congenital constriction band of the upper arm: the role of three-dimensional ultrasound in diagnosis, counseling and multidisciplinary consultation. Ultrasound Obstet Gynecol 2004;23:520–2.
Pedersen TK, Thomsen SG. Spontaneous resolution of amniotic bands. Ultrasound Obstet Gynecol 2001;18:673–4.
Quintero RA, Morales WJ, Phillips J, et al. In utero lysis of amniotic bands. Ultrasound Obstet Gynecol 1997;10:316–20.
Zimmer EZ, Bronshtein M. Fetal polydactyly diagnosis during early pregnancy: clinical applications. Am J Obstet Gynecol 2000;183(3):755–8. Internet resources
www.steps-charity.org.uk/
www.ponseti.info
Fetal abnormalities: head and neck Cystic hygroma
Definition
Cystic hygromas (CHs) are fluid-filled spaces that are typically noted at the back or sides of the fetal neck (Fig. 7.11.1). They may have septations within them and in the first trimester are usually manifest as increased nuchal translucency. A distinct group of cystic lesions that are sometimes identified in the third trimester tend to be fetal manifestation of lymphangiomas.
Incidence
Variable 1–3:1000.
Aetiology
The vast majority of CHs are thought to be associated with chromosomal abnormalities (>50%), most commonly Turner’s syndrome. Some authors consider CHs as an independent marker for aneuploidies, whereas others have questioned this by demonstrating a strong relationship with increased nuchal translucency. In the absence of aneuploidies, CH has still a high incidence of associated malformations of the fetal heart, skeletal system, etc. (>33%). More recently the association between CH and several genetic syndromes has been described, including Noonan’s syndrome, Cornelia De Lange syndrome and Roberts syndrome.
Management
The presence of CH in the first trimester should prompt a management plan as for increased nuchal translucency over the 95th centile. This should include offering invasive prenatal diagnosis, early fetal echocardiography, and a thorough search for other structural anomalies. Markers for specific genetic syndromes should be sought and the genetics team involved early in the process of counselling.
Prognosis
The outcome for CH in general is guarded and depends on the underlying diagnosis and associated abnormalities, if any. In the case of isolated CH, where no chromosomal, structural, or genetic associations have been established, the prognosis tends to be good in about 95% of cases. However, only a small proportion of CH is isolated. Fetal goitre
Definition
Fetal goitre is a diffuse enlargement of the fetal thyroid gland.
Incidence
Fetal goitre is a very rare abnormality and can occur in the presence of a hyperthyroid, hypothyroid or euthyroid state in the fetus.
Aetiology
Maternal hyperthyroid states due to autoimmune antibodies are at risk of causing fetal goitre. The risk is increased when the mother is on antithyroid drugs, as both the antibodies and the drugs can cross the placenta and cause goitre. This risk is present when mothers are euthyroid or hypothyroid following surgical or radioisotope treatment for Graves’ disease. Women in areas with endemic iodine deficiency or those with inborn errors of metabolism are similarly at risk of having fetal goitre.
Ultrasound findings
Typically, the fetal goitre is usually detected in the third trimester and is noted as a diffuse swelling in the anterior region of the fetal neck. The mass can be seen to be symmetrical on both sides of the fetal neck abutting the trachea (Fig. 7.11.2). Normograms are available for the length and width of the fetal thyroid and measurements above 2 SD are considered abnormal. The presence of goitre does not indicate hyper- or hypothyroid status in the fetus (this can only be diagnosed with fetal blood samples). In general hyperthyroid fetuses tend to have a higher fetal heart rate (over 160 bpm). If the goitre is big enough to obstruct the oesophagus, fetal swallowing may be hampered and polyhydramnios follows. Rarely, the goitre may be big enough to cause hyperextension of the fetal neck causing problems during labour.
Management
In the vast majority of cases the fetal goitre remains a chance finding, with the scan being done for other indications. Some centres have protocols to scan fetuses of mothers with thyroid abnormalities even in the euthyroid state. As the scan does not usually give a clue to the fetal thyroid state, fetal blood sampling may be required to clarify this. Most of the available literature is on case reports due to the rarity of the fetal condition. This makes counselling difficult and is individualized for each fetus. There are several case reports suggesting successful treatment of the fetus with thyroid hormones and antithyroid drugs. Polyhydramnios may require drainage. It would be prudent to consider delivery of the fetus with a goitre in a tertiary centre in the event of there being tracheal compression. Postnatal follow up is usually organized by the neonatal team for managing the thyroid status in the baby.

Fig. 7.11.1 Cross-section through the fetal head at the level of the occiput (a) and fetal neck (b) showing the cystic hygromas with septae.
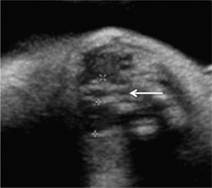
Fig. 7.11.2 Coronal section at the level of the fetal neck showing enlarged fetal thyroid gland (within callipers) on either sides of the fetal trachea (≥).

Fig. 7.11.3 Image showing cervical teratoma with mixed echogenicities.
Prognosis
Although there are no long-term studies, shrinkage of the goitre is the norm in the postnatal period, usually following treatment. Obviously congenital hypothyroidism is a potential problem if the condition is not appropriately treated. Cervical teratomas
Definition
Cervical teratomas are masses noted on either side of the fetal neck, usually having a mixture of cystic and solid components and derived from any/or all of the embryonic tissues.
Incidence
These masses are extremely rare and the precise incidence is difficult to assess.
Aetiology
Cervical teratomas are not associated with underlying fetal conditions and are almost entirely sporadic.
Ultrasound findings
The vast majority of these lesions are incidental findings when identified in the second trimester. However, in the third trimester, the presenting clinical feature may actually be polyhydramnios leading to this fetal diagnosis. Invariably, the mass is unilateral on the anterolateral aspect of the fetal neck with cystic and solid components (Fig. 7.11.3) extending to the face and crossing the midline. These lesions usually do not have high vascularity as they are essentially benign. Depending on the size of the mass and its extensions, there might be polyhydramnios and hyper-extension of the fetal neck. As these are isolated conditions, the rest of the fetal anatomy is usually normal.
Management
Antenatal management of these lesions include serial scans to assess the size of the lesion, fetal growth velocity and the development of polyhydramnios. In the event of there being polyhydramnios developing, amnio drainage should be considered with a view to reducing the risk of preterm delivery. A multidisciplinary team needs to be involved in the management of the perinatal period, comprising neonatologists, neonatal anaesthetists, paediatric ENT specialist, and a paediatric surgeon along with the obstetric team. This is in the event of the baby needing an ex utero intrapartum treatment (EXIT) procedure at delivery and emergency tracheostomy for establishing an airway. The baby will need surgery to remove the cervical mass postnatally and plastic surgery to try and restore the anatomy.
Prognosis
The prognosis for these lesions tends to be very good as they are isolated lesions and do not tend to recur. Further reading
Castillo F, Peiro JL, Carreras E, et al. The exit procedure (ex-utero intrapartum treatment): management of giant fetal cervical teratoma. J Perinat Med 2007;35:553–5.
Larsen ME, Larsen JW, Hamersley SL, et al. Successful management of fetal cervical teratoma using the EXIT procedure. J Matern Fetal Med 1999;8:295–7.
Martino F, Avila LF, Encinas JL, et al. Teratomas of the neck and mediastinum in children. Pediatr Surg Int 2006;22:627–34.
Sayan A, Karaçay S, Bayol U, Arikan A. Management of a rare cause of neonatal airway obstruction: cervical teratoma. J Perinat Med 2007;35:255–6.
Molina FS, Avgidou K, Kagan KO, et al. Cystic hygromas, nuchal edema, and nuchal translucency at 11–14 weeks of gestation. Obstet Gynecol 2006;107:678–83.
Kharrat R, Yamamoto M, Roume J, et al. Karyotype and outcome of fetuses diagnosed with cystic hygroma in the first trimester in relation to nuchal translucency thickness. Prenat Diagn 2006;26:369–72.
Malone FD, Ball RH, Nyberg DA, et al. FASTER Trial Research Consortium. First-trimester septated cystic hygroma: prevalence, natural history, and pediatric outcome. Obstet Gynecol 2005:106:288–94.
Goktolga U, Karasşahin KE, Gezginç K, et al. Intrauterine fetal goiter: diagnosis and management. Taiwan J Obstet Gynecol 2008;47:87–90.
Miyata I, Abe-Gotyo N, Tajima A, et al. Successful intrauterine therapy for fetal goitrous hypothyroidism during late gestation. Endocr J 2007;54:813–7.
Hashimoto H, Hashimoto K, Suehara N. Successful in utero treatment of fetal goitrous hypothyroidism: case report and review of the literature. Fetal Diagn Ther 2006;21:360–5.
Van Loon AJ, Derksen JT, Bos AF, Rouwe CW. In utero diagnosis and treatment of fetal goitrous hypothyroidism, caused by maternal use of propylthiouracil. Prenat Diagn 1995;15:599–604.
Polak M, Legac I, Vuillard E, et al. Congenital hyperthyroidism: the fetus as a patient. Horm Res 2006;65:235–42.
Fetal abnormalities: skeletal abnormalities/dysplasias Definition
The skeletal dysplasias or osteochondrodysplasias are a group of conditions affecting bone growth and development. However, they may be associated with other congenital anomalies.
There are over 370 forms that have been carefully classified into 37 groups based on the radiological and molecular abnormalities.
In addition, other genetic syndromes and chromosomal abnormalities may be associated with bone abnormalities and should be considered in the differential diagnosis. Ultrasound findings
Some of the lethal skeletal dysplasias may present at 11–14 weeks with increased nuchal translucency and/or fetal oedema with very short long bones.
Femur length is measured routinely during the anomaly ultrasound scan at 18–24 weeks gestational age. It is compared with the biparietal diameter (BPD) and abdominal circumference (AC) to check for proportionate growth. A disproportionately short femur at this stage may indicate:
• inaccurate dating
• a small normal baby
• early intrauterine growth retardation
• chromosomal disorder
• skeletal dysplasia
• a malformation syndrome.
In a recent retrospective study it was found that 40% of fetuses with isolated short femurs had severe intrauterine growth restriction (IUGR) associated with abnormal umbilical Doppler studies. This group had a high incidence of pre-eclampsia (36%) and intrauterine death (33%).
Occasionally, non-lethal forms may be identified incidentally by measuring femur length in the third trimester.
Isolated or asymmetrical limb abnormalities may also be identified at any stage.
Approximately 5% of fetus with short long bones had no radiological abnormalities after birth. Thus, the outcome can range from lethality to normality. Epidemiology
The skeletal dysplasias are individually rare, but as a group occur with an incidence of 1 in 5000 in the newborn period. The incidence is presumably higher antenatally as many are lethal in utero. They account for 5% of all genetic disorders. Of those detected in the antenatal period, approximately 70% are detected before 24 weeks and 30% after 24 weeks. Pathology
The appendicular and axial skeleton undergo endochondral ossification of a cartilage template from early human gestation. The skull, clavicles and mandible ossify via intramembranous ossification from 8 weeks’ gestation. Secondary (epiphyseal) ossification can be seen from 20 weeks’ gestation. Many of the genes involved have been identified and code for collagens and proteins involved in bone development. Aetiology
Skeletal abnormalities may be a feature of skeletal dysplasias, chromosomal disorders or malformation syndromes.
Skeletal dysplasias are a heterogeneous group. They may be inherited as autosomal dominant, autosomal recessive, or X-linked disorders. A few are associated with maternal disease or teratogens.
An accurate diagnosis is essential for accurate assessment of recurrence risks and prognosis.
The most important decision on identification of a skeletal dysplasia is whether or not the condition is likely to be lethal.
Lethal skeletal dysplasias
The three most common lethal skeletal dysplasias accounting for 40% for all the lethal dysplasias are:
• thanatophoric dysplasia (11%)
• osteogenesis imperfecta type II (20%)
• achondrogenesis type II (8%).
Thanatophoric dysplasia
This is a severe, sporadic, lethal skeletal dysplasia due to mutations in the fibroblast growth factor receptor 3 (FGFR3). Thanatophoric means ‘death bringing’, as the affected baby dies in the neonatal period due to the severe thoracic dystrophy. Antenatally, the fetus presents in the first or second trimester with marked shortening of all the long bones, relative macrocephaly, frontal bossing, absent nasal bone, and very short fingers. The bones are well mineralized but the femur is often curved like a telephone receiver (type 1 thanatophoric dysplasia). The vertebral bodies are small but present. Occasionally the baby is oedematous or hydropic and polyhydramnios develops in the third trimester.
The recurrence risk for this condition is very low (mode of inheritance: small thorax with short ribs, short limbs, and polydactyly. Some forms are associated with a median cleft lip. This group can be readily confused with Ellis–van Creveld syndrome and Jeunes asphyxiating thoracic dystrophy. Differentiation is important, as the latter are not always lethal in the neonatal period. The degree of shortening of the long bones and severity of the small thorax are more marked in the short rib polydactyly syndromes.
Perinatal or infantile hypophosphatasia
The most severe forms of hypophosphatasia present prenatally—possibly as early as 12 weeks. There is reduced bone mineralization of long bones and skull. Fractures may be present. The long bones are short. Bony spurs may be seen on the tibiae—a helpful diagnostic feature. This is an autosomal recessive condition due to mutations in tissue the non-specific alkaline phosphatase gene (TNSALP). The recurrence risk is therefore 25%.
Campomelic dysplasia
This condition is characterized by disproportionate short long bones presenting in the second trimester. The scapulae, if visualized, are hypoplastic and there is often bowing (campomelia) of the long bones especially the femurs. Three-quarters of karyotypic males have ambiguous genitalia or complete sex reversal. New sporadic mutations in SOX9 are the cause of this condition, but the recurrence risk is approximately 5% because of the incidence of gonadal mosaicism. Non-lethal skeletal dysplasias
The most common non-lethal skeletal dysplasias presenting antenatally are discussed below.
Achondroplasia
This is an autosomal dominant disorder but with an 80% new mutation rate so there is frequently no family history. The fetus may present in the third trimester with short long bones (particularly the proximal long bones) below the 5th centile in the third trimester. It is very unusual to see any reduction in bone length before 24 weeks’ gestation. Other ultrasound findings include, frontal bossing, relative macrocephaly, depressed nasal bridge, and the ‘trident’ hands with short fingers.
This condition is due to one of two point mutations in FGFR3 in 99% of cases (this is the same gene associated with thanatophoric dysplasia, which is due to different mutations), therefore the diagnosis may be confirmed antenatally by molecular analysis of amniotic fluid, fetal blood, or chorionic villus sampling (placental biopsy). This particular genetic test is quick and cheap and therefore useful during the pregnancy.
Achondroplasia is associated with severe short stature, and complications include deafness due to recurrent otitis media, hydrocephalus, and acute onset of spinal stenosis resulting in paraplegia. Intelligence is normal.
Spondyloepiphyseal dysplasia congenita
Spondyloepiphyseal dysplasia congenita (SEDC) is a rare autosomal dominant genetic bone dysplasia due to new dominant mutations in collagen II. It may be diagnosed by antenatal ultrasound in the second or third trimester with disproportionate short long bones and normal-sized thorax. However, it is frequently missed until early childhood.
This condition is associated with normal intelligence but extreme short stature and many orthopaedic complications including progressive scoliosis, hip dysplasia and premature osteoarthritis. The baby may have micrognathia and cleft palate. There is a significant risk of myopia and hearing loss. The recurrence risk is very low.
Osteogenesis imperfecta
There are several forms of osteogenesis imperfecta (OI). As previously mentioned, type II is the most severe, lethal form.
Type III is also severe but compatible with survival. Antenatal ultrasound scans may detect fractures of the long bones in the second or third trimester. However, although the long bones may be mildly shortened, they are not broad and crumpled as seen in type II. Again there may be reduced mineralization of the skull and some rib fractures. Some of these babies die in the neonatal period but survivors suffer from recurrent fractures due to minimal trauma and as a result are confined to a wheelchair. Intelligence is normal.
Types I and IV are the mildest forms and usually present in childhood but may present with one or two fractures in the fetus or neonate. The long bones may be slightly shortened. There is often an autosomal dominant family history of an increased frequency of fractures.
All four types of OI are due to different dominant mutations in the α-1 and α-2 chains of collagen 1. Types I and IV may be inherited from an affected parent, but types II and III are not inherited as it is unlikely for affected individuals to reproduce. There are rare autosomal recessive forms and there is a risk of gonadal mosaicism—overall the recurrence risk is 5–7%. Others to consider
Jeunes asphyxiating thoracic dystrophy and Ellis–van Creveld syndrome
These rare autosomal recessive conditions may present in the second trimester with disproportionate long bones and narrow thorax. Polydactyly is also a feature, so they can, occasionally, be confused with the short rib polydactyly group (see above). Ellis van Creveld may be associated with congenital heart disease. Jeunes syndrome may be fatal in the neonatal period and survivors may develop chronic renal failure. Ellis–van Creveld syndrome is less frequently lethal. Recurrence risk for both is 25%.
Chondrodysplasia punctata
Chondrodysplasia punctata (CDP) is a very heterogeneous group of conditions associated with premature ossification of the epiphyses presenting as stippling, which can sometimes be seen on a careful ultrasound examination and absent nasal bone. This can be inherited in a X-linked dominant (Conradi–Hunerman–Happle syndrome, marked asymmetry, girls only), X-linked recessive (brachytelephalangic CDP, boys only, less severe), autosomal recessive (rhizomelic chondrodysplasia punctata associated with marked developmental delay), maternal illness (systemic lupus erythematosus, maternal hyperemesis) and teratogens (warfarin). Recurrence risk and prognosis depends on the specific diagnosis (Irving et al. 2008). Prognosis/recurrence risks
The prognosis and recurrence risk depends on the type of skeletal dysplasia. The lethal skeletal dysplasias should be identifiable in the antenatal period. These fetuses frequently die in utero or in the neonatal period. In a retrospective study by Krakow et al (2008), the correct diagnosis was made in only 42%, however in the same publication, lethality was accurately predicted in 96.8%.
Correct diagnosis is essential for prediction of complications. Many of the surviving skeletal dysplasias are associated with orthopaedic complications and morbidity.
The recurrence risk is also based on the correct diagnosis as the mode of inheritance varies depending on the type of dysplasia. Clinical approach
History
A detailed three-generation family history can be helpful in making a specific diagnosis. Parental heights should be determined.
Consanguinity suggests an autosomal recessive disorder Increased paternal age is associated with an increased incidence of achondroplasia and Apert’s syndrome.
Increased maternal age is associated with increased risk of chromosomal aneuploidy.
Pregnancy history
Disproportionate short stature may be associated with early placental insufficiency so early bleeding or the loss of a twin may be significant.
Drug exposure exposure to warfarin (CDP), sodium valproate (isolated limb defects).
Maternal illness: hyperemesis resulting in vitamin K deficiency or maternal systemic lupus erythematosis (CDP).
Maternal diabetes can be associated with sacral agenesis, vertebral anomalies and asymmetrical shortening of the long bones particularly the femurs. Examination
Assessment of viability
Most lethal skeletal dysplasias present before the third trimester. The following should be considered indicators of lethality:
• early presentation
• very short long bones (femur length/AC ratio dystrophy/high cardiothoracic ratio
• hydrops fetalis or oedema
• very poor mineralization of the long bones and/or skull. The converse is also true, the condition is unlikely to be lethal if
• the disproportion presents in the third trimester
• the thorax looks a normal size
• there is no oedema of the fetus.
Aids for making a specific diagnosis
Long bones
Measurement of all long bones/degree of disproportion.
Pattern of shortening
• Rhizomelia: proximal part (achondroplasia/CDP)
• Mesomelia: middle part
• Acromelia: distal part.
Bone modelling
• Bowing (campomelic dysplasia/OI)
• fractures (OI/hypophophatasia)
• absence
• bone density/mineralization (decreased in OI/hypophosphatasia/achondrogenesis: increased in sclerosing bone disorders)
• epiphyseal stippling (chondrodysplasia punctata)
• asymmetry (OI/maternal diabetes/isolated limb defects)
• scapulae (small/absent in campomelic dysplasia).
Other measurements
• BPD (relatively increased achondroplasia/thanatophoric)
• Abdominal circumference (IUGR)
• Chest circumference (cardiothoracic ratio, thoracic/AC ratio for assessment of lethality, fractures of ribs—OI/hypophosphatasia).
Skull
• Mineralization (reduced in OI type II/hypophosphatasia/achondrogenesis)
• cloverleaf (thanatophoric dysplasia type II)
• craniosynostosis (see Chapter 7.7, Genetic disorders).
Vertebrae
• Absent mineralization of vertebral bodies (achondro-genesis type II/hypophosphatasia)
• scoliosis
• hemivertebrae.
Hands and feet
• Brachdactyly (short fingers)/trident hands (achondroplasia and thanatophoric dysplasia)
• extra fingers (polydactyly) (short rib polydactyly syndromes, Ellis–van Creveld and Jeunes asphyxiating thoracic dystrophy)
• hitchhiker thumb (diastrophic dysplasia)
• radial aplasia (trisomy 18, fanconi anaemia, Holt Oram syndrome, TAR syndrome)
• syndactyly: soft tissue or bony fusion (Apert’s syndrome)
• talipes equinovarus (diastrophic dysplasia, campomelic dysplasia, SEDC).
Facial profile
• Micrognathia (achondrogenesis and SEDC)
• median cleft lip (short rib polydactyly)
• absent nasal bridge (chondrodysplasia punctata).
Genitalia
• Abnormalities of the genitalia or sex reversal (campomelic dysplasia, Beemer Langer syndrome (form of short rib-polydactyly))
• hypospadias (intrauterine growth retardation). Investigations
Chromosome analysis
Chorionic villous sample, amniocentesis or fetal blood depending on gestation. Chromosomal disorders including trisomy 21 may present with short femurs, although an isolated short femur is a poor predictor for trisomy 21.
Molecular analysis
May be usefully performed for achondroplasia during the pregnancy. If there is a family history of a known skeletal dysplasia, the causative mutation can often be identified prior to the pregnancy and prenatal diagnosis can be offered at 11 weeks’ gestation by CVS.
In most other skeletal dysplasia, most genetic tests take too long to be of practical use during the pregnancy, but stored DNA can be very helpful in making a specific diagnosis after birth. Counselling
Lethal disorders
Assessment of the likelihood of lethality is essential for the further management of the pregnancy. Termination of pregnancy is an option throughout the pregnancy for a lethal disorder. Post-mortem examination and DNA storage should be encouraged, as a specific diagnosis helps to determine accurate recurrence risks. If a post-mortem is refused, the couple should be encouraged to allow a skeletal survey and external examination as it is often possible to make a diagnosis based on these alone.
Non-lethal disorders
Counselling is more difficult in non-lethal disorders. These are frequently diagnosed later in the pregnancy. Often it is difficult to make a precise diagnosis until the baby is born. Parents should be warned that it may be a few years before a specific diagnosis is made. For this reason it is helpful to involve a geneticist who can organize investigations after birth and follow up on a regular basis. Disproportionate short limbs are often easier to detect on antenatal ultrasound than plain radiographs after birth because of the availability of antenatal centile charts.
The complications also vary depending on the specific diagnosis. However, as a general rule, if chromosomal abnormalities are excluded, most of the surviving skeletal dysplasias are associated with normal intelligence.
Prognosis, final height, and recurrence risk depends on the underlying cause of the skeletal dysplasia. Orthopaedic complications are frequent in this group.
It is important to be aware that early growth retardation may present as disproportionate short long bones. Management
• Correct assessment of viability based on ultrasound findings (as above).
• Assessment of placental function and abdominal circumference to exclude IUGR.
• Attempt at making a specific diagnosis based on history and ultrasound findings (this is only possible in less than 50% of cases).
• Exclusion of chromosomal abnormality by CVS/amniocentesis or fetal blood.
• Molecular analysis of FGFR3 if achondroplasia is suspected.
• Serial scans to assess fetal growth.
• If the baby is suspected of having osteogenesis imperfecta, a normal vaginal delivery is still the preferred mode of delivery unless there is any indication that the delivery will be difficult.
Involvement of
• Clinical geneticists for arrangement of further testing and follow-up.
• Neonatologists
• Paediatric orthopaedic surgeon.
Careful follow-up
After intrauterine death or termination of pregnancy:
• post-mortem examination
• radiology: full skeletal survery
• store DNA.
After live birth
• Careful clinical examination by neonatologist and/or geneticist
• Skeletal survey at birth with expert radiological opinion
• The baby will require follow up by paediatrician/orthopaedic surgeon/clinical geneticist
• Depending on diagnosis may require hearing and ophthalmic assessment on a regular basis
• If no specific diagnosis is made—annual follow-up by the clinical geneticist is suggested with repeat skeletal survey at the age of 3 years.
Genetic counselling
To discuss
• prognosis
• recurrence risks
• prenatal diagnosis in future pregnancies by genetic testing or detailed scans. Further reading
Firth HV, Hurst JA. Oxford Desk reference: clinical genetics. Oxford: Oxford University Press 2003.
Irving MD, Chitty LS, Mansour S, Hall CM. Chondrodysplasia punctata: a clinical diagnostic and radiological review. Clin Dysmorphol 2008;17:229–41.
Krakow D, Alanay Y, Rimoin LP, Lin V, Wilcox WR, Lachman RS, Rimoin DL. Evaluation of prenatal-onset osteochondrodysplasias by ultrasonography: a retrospective and prospective analysis. Am J Med Genet A 2008; 1(146A): 1917–24.
Mansour S, Hall CM, Pembrey ME, Young ID. A clinical and genetic study of campomelic dysplasia. J Med Genet. 1995;32:415–20.
Papageorghiou AT, Fratelli N, Leslie K, et al. Outcome of fetuses with antenatally diagnosed short femur. Ultrasound Obstet Gynecol 2008;31:507–11.
Superti-Furga A, Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A. 2007;143:1–18. Internet resources
The skeletal dysplasia group are a UK association created for the promotion of teaching and research of the skeletal dysplasia. Their publications include summaries of the skeletal dysplasias: www.skeletaldysplasiagroup.org.uk/index.html
Some of the summaries may be accessed on the companian web site for the Oxford Desktop Reference—Clinical Genetics (Firth and Hurst): www.oup.com/uk/booksites/content/0192628968/
Further information for patients for specific disorders can be obtained on the Contact a Family website: www.cafamily.org.uk/index.html Patient resources
Restricted Growth Association (UK), PO Box 4008, Yeovil BA20 9AW. Tel: 01935 841364 (Mon, Wed and Thur 9 am–5 pm; Tues 9 am–9 pm; Fri 9 am–12 noon); Fax: 01935 841364; e-mail: office@restrictedgrowth.co.uk: www.restrictedgrowth.co.uk
Little People of America, Inc. (USA), 250 El Camino Real, Suite 201, Tustin, CA 92780; Toll-free: (888) LPA-2001 (English and Spanish); Direct: (714) 368 3689; Fax: (714) 368 3367; E-mail: info@lpaonline.org
Antenatal Results and Choices ARC (UK), 73 Charlotte Street, London W1T 4PN; Admin: 0207 631 0280; Helpline: 0207 631 0285; E-mail: info@arc-uk.org
Fetal abnormalities: thorax Congenital cystic adenomatous malformation
Definition
A congenital cystic adenomatous malformation (CCAM) is a benign condition characterized by an abnormal mass of lung tissue, located usually on one lobe of the fetal lung. This lesion derives its blood supply from the pulmonary vasculature. It belongs to a heterogeneous group of echogenic lesions of the fetal lung that includes CCAM, pulmonary sequestration, bronchial atresia and transient bronchial obstruction. All of them can present antenatally as an echogenic fetal lung lesion. The commonest of these is CCAM.
Incidence
CCAM is thought to occur in approximately 1 in 4000 pregnancies.
Aetiology
The exact cause for this abnormality is unknown. It is thought to occur as a consequence of arrested and abnormal growth of the terminal respiratory bronchioles. It is usually an isolated finding and does not have a genetic or chromosomal basis. It may be occasionally associated with heart or renal abnormalities.
Ultrasound features
CCAM presents as an intrathoracic cystic or solid echogenic lesion (Fig. 7.13.1) that is usually unilateral and more commonly affects the lower lobe of the fetal lung. Both sides of the fetal lung and both sexes are affected equally. As this does not function as normal lung tissue, many fetuses show obvious cystic areas, which can cause a significant mediastinal shift. Antenatally these lesions are classified as macrocystic (with obvious cysts), microcystic (cysts not visible to the naked eye, lesion appears echogenic) or mixed. A thorough fetal anatomical survey is essential to rule out coexisting anomalies. A detailed survey of the blood supply to this lesion should be sought in order to differentiate this from the even rarer pulmonary sequestration. The latter derives its blood supply from the dorsal aorta and could lead to a hyperdynamic circulatory state owing to the shunting from this lesion. This lesion can be difficult to differentiate from congenital high airway obstruction, which causes trapping of the secretions in the lung and is imaged as an echogenic lobe or the entire lung. Transient blockage of the bronchial tree with a mucus plug tends to resolve spontaneously over the course of the pregnancy, but bronchial atresia tends to persist.
Serial scans should be organized in order to monitor the fetus for signs of fetal hydrops as that would dramatically alter the outcome for the pregnancy. Additionally, progressive increase in the size of the cysts might require thoraco-amniotic shunting.

Fig.7.13.1 Transverse section of the fetal thorax showing an echogenic lesion in the right side of the fetal chest, with mediastinal shift pushing the heart to left side.
Management
Antenatal
The vast majority of fetuses with CCAM have an uneventful course. Occasionally, intervention may need to be offered as mentioned above. It is important that the prospective parents are provided with an opportunity to meet the medical and surgical team who will be looking after the baby postnatally. It is also important to make a plan for postnatal management.
Bilateral disease and hydrops fetalis are indicators of poor outcome, whereas mediastinal shift, polyhydramnios and early detection are not poor prognostic signs. Serial scans have shown spontaneous reduction of these lesions with difficulty in demonstrating these lesions as the baby may ‘outgrow’ these lung lesions. This should be interpreted with caution as lesions that have regressed or disappeared antenatally may still need surgery postnatally.
Postnatal
Postnatal management includes chest X-ray and CT scan to identify the site and size of the lesion. The urgency of these investigations is based on the presence of symptoms in the newborn. Most lesions are removed surgically in order to eliminate the possibility of infection and malignancy if left in situ. Surgery can be either through an open thoracotomy or a minimally invasive thoracotomy.
Prognosis
The outcome in general for the uncomplicated CCAMs is very good with the vast majority of infants growing and developing normally. Congenital diaphragmatic hernia
Definition
Congenital diaphragmatic hernia is an anomaly in the diaphragm that does not develop normally allowing the abdominal contents to herniate into the thoracic cavity. This results in poor lung development.
Incidence
CDH is thought to occur in approximately 1 in 2500 pregnancies.
Aetiology
Essentially, the defect in the development of the lung is likely to be due to defective mesenchymal incorporation. Several conditions are thought to result in this maldevelopment, which includes chromosomal abnormalities (trisomies 13, 18 and 21), genetic syndromes (Fryn’s syndrome), and deficiency in retinoic acid.
Ultrasound features
Left-sided CDH (Fig. 7.13.2a) is usually identified at the 18–22-week scan when the fetal stomach and bowel are noted in the chest along with a mediastinal shift to the right. Identification can be possible at the 11–14-week scan, when the condition is often associated with increased fetal nuchal translucency. Rarely, the liver is noted in the chest in cases of right-sided hernia (Fig. 7.13.2b) and these can be more difficult to diagnose.
Fig. 7.13.2 (a) Transverse section through the fetal thorax showing the stomach bubble in the left side of the thorax, pushing the fetal heart to the right hemithorax: left-sided diaphragmatic hernia.

Fig. 7.13.2 (b) Transverse section through the fetal thorax showing the fetal liver in the right side of the thorax, pushing the fetal heart to the left hemithorax: Right-sided diaphragmatic hernia. Area within callipers shows normal lung tissue.
CDH is an isolated finding in about 60% of cases. It can be present in association with cardiac abnormalities, other markers for chromosomal aberrations, and growth restriction. A thorough search for other abnormalities should be made and options of invasive diagnosis discussed with the parents. A proportion of CDHs may go undiagnosed, as not all hernias present at the time of the anomaly scan.
CDHs have to be differentiated from eventration of the diaphragm. This can sometimes be difficult as the thin diaphragm in the latter may not be visible and a diagnosis of CDH is made.
Management
Antenatal
The management of CDH is hinged on early inclusion of a multidisciplinary team, including neonatologists, paediatric surgeon, geneticist, and the fetal medicine team. It is important to provide as much details of outcome to the prospective parents as possible from all angles so that they are able to make an informed decision about the pregnancy. The initial management is geared towards making a diagnosis of the extent of involvement and if possible, the underlying aetiology. The pregnancy is then followed up with serial scans assessing fetal wellbeing and the development of polyhydramnios. Plans must be in place to deliver the baby in a tertiary centre that offers neonatal surgery for the CDH.
Several features have been reported as predictors of outcome in antenatally diagnosed CDH. The lung-to-head ratio (LHR), which measures the remaining area of normal lung in relation to the fetal head circumference measured on ultrasound and fetal lung volume measured with fetal MRI scan, are considered to have a role in predicting outcome in CDH. In addition, the presence of the liver in the chest confers a poor prognosis. These fetuses may be considered for in utero procedures such as tracheal occlusion using a balloon or plug to promote lung growth and development. This is reported to improve survival rates in some babies, but the main drawback is preterm delivery due to the invasive intervention, and the procedure is currently a subject of a randomized trial.
Postnatal
Planned delivery in a tertiary centre is required. Once the newborn has been stabilized, surgical repair of the diaphragmatic defect is carried out. In most cases it is closed by primary repair, although some cases may require closure with a patch. Extensive respiratory support is required, including ventilation and measures to deal with the pulmonary hypertension, such as the use of nitric oxide. Pulmonary hypoplasia, abnormal pulmonary vasculature, and lung injury secondary to mechanical ventilation are the main causes of both mortality as well as long-term respiratory morbidity. A large proportion of these babies have long-term morbidity affecting gastrointestinal and neurological development as well.
Prognosis
The outcome in babies with CDH depends on the size and content of the hernia and also the associated structural chromosomal or genetic conditions. In general, for a left-sided hernia with just bowel and no liver in the fetal chest, the survival rate varies from about 40% to 60% and there is a much higher rate of other morbidity. Fetal pleural effusion
Definition
Accumulation of fluid in the fetal pleural cavity resulting in pleural effusion can be isolated or occur with fetal hydrops.
Incidence
Pleural effusion is thought to occur in 1 in 10 000 fetuses.
Aetiology
Pleural effusion may occur as an isolated finding or in association with other conditions. Such secondary effusions occur along with diaphragmatic hernia and other lesions compressing the lungs and mediastinum such as congenital cystic adenomatoid malformation and bronchopulmonary sequestration, mediastinal tumours, and cardiac malformations. It is also noted with chromosomal abnormalities, congenital infections such as parvovirus B19, which cause fetal hydrops, and genetic syndromes such as Noonan’s syndrome.
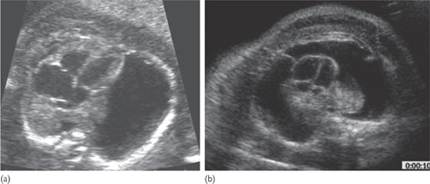
Fig. 7.13.3 (a) Transverse section through the fetal thorax showing pleural effusion (anechoeic area) in the right hemithorax with significant mediastinal shift to the left. (b) Transverse section through the fetal thorax showing bilateral pleural effusions (anechoeic area) in a fetus with hydrops (note the skin oedema). Both fetal lungs are hypoplastic.
Ultrasound features
A fluid-filled space surrounding one (Fig. 7.13.3a) or both fetal lungs (Fig. 7.13.3b) in isolation is seen and this may be associated with ascites, skin oedema, or pericardial effusion, suggesting fetal hydrops. A detailed fetal survey with specialist fetal echocardiography is essential to rule out possible associations, and the laboratory work-up should include screening for maternal viral infections, antibodies, and an invasive test for karyotyping. In the vast majority of cases with isolated effusion, no obvious cause can be identified. Accumulation of fluid in the pleural space may lead to pulmonary hypoplasia, compression of the heart, and obstruction of venous return with subsequent development of hydrops and compression of the oesophagus leading to polyhydramnios.
Management
Antenatal
Antenatal management of fetal pleural effusion should include serial scans and counselling involving the neonatal, genetics, and the fetal medicine team. The main consequence of the pleural effusion is that of pulmonary hypoplasia and pulmonary vascular hypertension. The risks of developing these depend on the time and duration of the pleural effusion. Worsening effusion indicates impaired pulmonary development and this should prompt the discussion of options to drain the effusion either by thoracocentesis or by a thoraco-amniotic shunt. Both procedures are not without risk, and this needs to be balanced against the high risk of mortality and morbidity following pulmonary hypoplasia. The delivery should be at a centre that can offer tertiary level neonatal care.
Postnatal
Postnatal care aims to stabilize the baby by promoting lung expansion (pleural drains may be needed) and by reducing the pulmonary vascular resistance. Respiratory dysfunction combined with prematurity are the main factors that contribute to neonatal death.
Prognosis
Fetal pleural effusions may regress spontaneously or show rapid deterioration with onset of fetal hydrops. There are no indicators to suggest the course of the effusion in a given fetus. The outcome is mainly determined by any underlying fetal condition/abnormality, and also depends on the trend in the fluid collection, the gestational age of occurrence, occurrence of polyhydramnios and the gestational age of delivery. Fetuses with hydrops tend to show a very high mortality of about 60% in the perinatal period. Fetuses with isolated pleural effusion that show spontaneous regression do very well. Survival for fetuses with persistent effusions following in utero intervention appears better for those without hydrops (around 80%) than those presenting with hydrops (around 60%). Further reading
Cavoretto P, Molina F, Poggi S, et al. Prenatal diagnosis and outcome of echogenic fetal lung lesions. Ultrasound Obstet Gynecol 2008;32:769–83.
Jani JC, Nicolaides KH, Gratacos E, et al. Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol 2009;34:304–10.
Lakhoo K. Management of congenital cystic adenomatous malformations of the lung Arch Dis Child Fetal Neonatal Ed 2009; 94; F73–6.
Maeda H, Shimokawa H, Yamaguchi Y, et al. The influence of pleural effusion on pulmonary growth in the human fetus. J Perinat Med 1989;17:231–4.
Mann S, Wilson RD, Bebbington MW, et al. Antenatal diagnosis and management of congenital cystic adenomatoid malformation. Semin Fetal Neonatal Med 2007;12:477–81.
Rustico MA, Lanna M, Coviello DS, et al. Fetal pleural effusion. Prenat Diagn 2007;27:793–9.
van den Hout L, Sluiter I, Gischler S, et al. Can we improve outcome of congenital diaphragmatic hernia? Pediatr Surg Int 2009;25:733–43.
Fetal abnormalities: urinary system
Definition
Abnormalities of the genitourinary tract may affect the kidney alone, including irregularities of number, structure and location. Urinary tract dilatation can result from obstruction or reflux and occur at any level, with or without associated with renal dysplasia.
Epidemiology
Genitourinary tract anomalies are relatively common, accounting for approximately 20% of all fetal malformations. The number detected antenatally varies widely, from 1 in 70 to 1 in 1200 across different centres internationally. The variation is likely to be due to gestational age at scan and definitions used rather than true population differences.
Pathology
Renal
• Renal agenesis may be unilateral or bilateral; it results from early degeneration of the ureteric bud or failed interaction between the ureteric bud and the blastema. Unilateral renal agenesis occurs in 1:3000 pregnancies. It may occur in isolation, in association with minor abnormalities such as uterine malformations, or more complicated structural abnormalities such as the VACTERL sequence:
• Vertebral anomalies
• Anal atresia
• Cardiovascular anomalies
• Tracheoesophageal fistula
• Esophageal atresia
• Renal (kidney) and/or radial anomalies
• Limb defects.
• Duplex kidney is more common in females and arises from premature division of the ureteric bud resulting in two pelvicalyceal systems. The upper ureter usually enters the bladder abnormally, or may form ectopic connections to other pelvic organs. It may be associated with obstruction or reflux.
• Renal ectopy may involve one or both kidneys. It affects 1 in 1200 pregnancies and results in a caudally displaced kidney, which if located in the pelvis carries a risk of obstruction.
• A horseshoe kidney occurs due to fusion of the kidneys, usually at the lower pole. Function is usually normal but it may be associated with aneuploidy (i.e. monosomy X).
• Autosomal recessive polycystic kidney disease (infantile polycystic kidney disease) results in large kidneys which retain their shape but contain multiple cysts, often associated with other anomalies.
• Autosomal dominant polycystic kidney disease (adult polycystic kidney disease) may have an early and severe expression and therefore be detected in utero.
• Multicystic dysplastic kidney is an idiopathic condition in which the kidney loses its shape due to the presence of multiple cysts. The ureter is usually atretic.
Urinary tract
• Ureteric junction obstruction is the commonest abnormality of the fetal urinary tract. It may rarely be caused by external compression from an ectopic vessel.

Fig 7.14.1 Ultrasound image showing ureterocele.
• Megaureter occurs as a result of dysfunction of the uterovesical junction.
• Ureterocele is a cystic dilatation of the intravesical ureter. It arises from an ectopic location of the ureter within the bladder. It may obstruct the corresponding ureter, and, if large, the bladder neck (Fig. 7.14.1).
• Obstruction of the bladder outlet (lower urinary tract obstruction) usually occurs within the urethra. This results in hypertrophy of the bladder wall and may also be associated with hydronephrosis and renal dysplasia. The commonest cause in males (1 in 5000–8000 boys) is posterior urethral valves; folds of mucosa along the posterior wall of the urethra resulting in a narrow lumen. Urethral atresia may also occur.
• Complex cloacal anomalies occur after failure of the urogenital sinus to divide normally, resulting in vesicovaginal or vesicourethral fistulae in females and urethrorectal fistulae in males.
• Urachal diverticulum results from failure of closure of the infrafunicular portion of the urachus, leading to a cystic mass within the cord wall which may persist as a vesicocutaneous fistula neonatally.
• Vesicoureteric reflux results in functional dilatation of the ureter and calyceal systems. Ultrasound findings
The fetal bladder may be imaged from the late first trimester and the fetal kidneys by 11–14 weeks. The sonographic corticomedullary differentiation of the kidneys takes place by 24 weeks. It is not always possible to distinguish between obstruction and reflux sonographically, or to determine the underlying pathology.
Renal
• Both forms of polycystic kidney disease may appear as bilateral enlarged kidneys with a hyperechogenic cortex (Fig. 7.14.2).
• Multicystic dysplastic kidneys are easily differentiated from polycystic kidneys sonographically due to the presence of macrocysts (0.5–3 cm) (Fig. 7.14.3).
Urinary tract
• Pyelectasis is renal pelvic dilatation without calyceal involvement and is found in 2% of pregnancies. It is defined as an anteroposterior (AP) diameter greater than 6 mm in the second trimester and 8–10 mm in the third. In most cases it is physiological but it may represent reflux or obstruction.
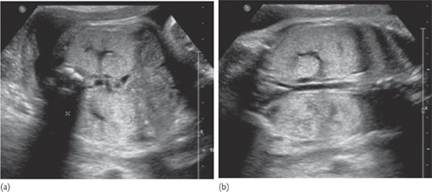
Fig 7.14.2 Bilateral echogenic kidneys in autosomal recessive (infantile) polycystic kidney disease in (a) axial and (b) coronal views.
• Fetal hydronephrosis is defined as an AP diameter greater than 15 mm and/or calyceal involvement (Fig. 7.14.4).
• Hydroureter appears as a convoluted transonic image between the kidney and bladder. A normal ureter is not visualized.
• A dilated bladder that does not empty over the course of the scan is suggestive of lower urinary tract obstruction. The bladder appears round, the wall may be hypertrophic (>3 mm). In the case of posterior urethral valves the dilated proximal urethra forms the ‘keyhole’ sign (Fig. 7.14.5). Aetiology
Overall, 12% of fetal urinary tract anomalies are associated with a chromosomal abnormality. Rare familial cases of renal agenesis have been reported, with variable patterns of inheritance. Cystic kidneys may occur with several multiple malformation conditions, for example the auto-somal recessive Meckel–Gruber syndrome. Prognosis
The prognosis for isolated unilateral renal lesions is good. For bilateral lesions or obstruction, the prognosis depends on an assessment of renal function. It is not yet established whether obstructive uropathy causes renal dysplasia, or the renal abnormality arises from a simultaneous disordered embryological process.
An assessment of fetal renal function may be made via
• measurement of liquor volume: fetal urine is the main source of amniotic fluid production in the second and third trimesters. Severe oligohydramios, particularly from 16–22 weeks’ gestation, results in pulmonary hypoplasia, which is invariably lethal. Anhydramnios also may result in fetal demise secondary to cord compression. Oligohydramnios is associated with a higher incidence of postnatal renal failure and perinatal mortality
• appearance of the fetal kidneys: hyperechogenicity of the cortex and the appearance of macrocysts are suggestive of renal dysplasia and poor renal function. However, the renal pelvic diameter is not a prognostic indicator; there may be gross enlargement and a thin cortex with normal renal function

Fig 7.14.3 Multicystic dysplastic kideney.

Fig 7.14.4 severe hydronephrosis with calyceal involvement.
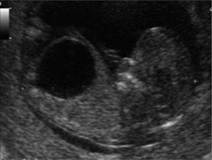
Fig 7.14.5 Fetal megacystis in the first trimester.
• fetal urinalysis: this has a controversial role in the assessment of renal function. An elevated urinary sodium and calcium show some correlation with poor postnatal renal function, but no single analyte or threshold is predictive.
No single measurement or combination of the above has been shown to accurately predict renal failure. Clinical approach
Diagnosis
History: key points
• The majority of urinary system abnormalities will be detected by routine antenatal scanning.
• A family history of an inherited condition, chromosomal anomaly or urinary tract defect should be considered when interpreting the scan findings.
Investigations: key points
• The diagnosis should be confirmed with a detailed ultrasound performed by a fetal medicine specialist. This should include an assessment for the presence of associated anomalies.
• A fetal karyotype should be considered. In isolated pyelectasis, due to the frequency of this finding in normal pregnancy, the likelihood of aneuploidy is low and invasive testing therefore not routinely performed. If there are associated abnormalities the risk of aneuploidy increases 10-to 20-fold.
Management Conservative: key points
• Unilateral renal lesions generally have a good prognosis and can be managed conservatively.
• In cases of bilateral hydronephrosis or features suggestive of lower urinary tract obstruction, conservative management, with regular ultrasound to monitor the condition, may be advised if the urinary tract dilatation is mild, or detected for the first time at a late gestation.
• Parents with a severe abnormality and poor prognosis may opt for conservative management.
• All parents, regardless of prognosis, should be referred to the paediatric team to discuss postnatal management, and delivery planned in a unit with appropriate neonatal care facilities.
Termination of pregnancy: key points
• In the case of a urinary tract anomaly with a poor prognosis termination of pregnancy may be offered.
• This is a difficult decision, particularly with the uncertainty in predicting renal function, and parents should be given ample time to discuss the options with the fetal medicine and paediatric teams.
• All parents who undergo termination of pregnancy or miscarry should be advised to have a post-mortem examination; this can confirm the antenatal findings and enable improved counselling for future pregnancies.
Fetal therapy: key points
• Various techniques have been proposed to relieve lower urinary tract obstruction; restoration of liquor volume allows pulmonary development and relief of the urinary pressure has been purported to allow normal renal development.
• Open fetal surgery carries a high risk of fetal and maternal morbidity and is not currently practised.
• Fetal endoscopy is difficult, is only applicable to posterior urethral valves and carries and increased risk of damage to associated organs through the use of laser in a small fetus.
• The use of a vesico-amniotic shunt, a double pigtail catheter inserted transabdominally, has been used more commonly. However, the evidence of benefit is insufficient, and there are associated risks of miscarriage, maternal, and fetal infection and injury.
• In the UK, the National Institute for Health and Clinical Excellence (NICE) therefore recommend that shunts are only used within the context of a clinical trial.
• The PLUTO trial is a multicentre randomized controlled trial to determine the effect of shunting and is now recruiting.
Follow-up
Postnatal management should involve radiological imaging of the urinary tract: further intervention will depend on the abnormality and renal function.
Future pregnancy planning
If a karyotypic abnormality or inherited disorder is present referral to a geneticist should be made. Further reading
Twining P. Uninary tract abnormalities. In: Twining P, McHugo J, Pilling D. Textbook of fetal abnormalities. London: Churchill Livingstone 2000: 269–314
Zaccara A, Giorlandino C, Mobili L, et al. Amniotic fluid index and fetal bladder outflow obstruction: do we really need more? J Urol 2005;174:1657–60.
Hutton KA, Thomas DF, Davies BW. Prenatally detected posterior urethral valves: qualitative assessment of second trimes-ter scans and prediction of outcome. J Urol 1997; 158; 1022–5.
Morris RK. Quinlan-Jones E, Kilby M, et al. Systematic review of fetal urine analysis to predict poor postnatal renal function in cases of congenital lower urinary tract obstruction. Prenat Diagn 2007;27:900–11.
Clark TJ, Martin WL, Divakaran TJ, et al. Prenatal bladder drainiage in the management of fetal lower urinary tract obstruction: a systematic review and meta-analysis. Obstet Gynecol 2003;102:367–82.
Dommergues M, Daikha-Dahmane F, Mueller F, et al. Kidney and urinary tract disorders. In: Rodeck CH, Whittle MJ (ed.) Fetal medicine basic science and clinical practice. London: Churchill Livingstone 2008: Ch 35. Internet resources
PLUTO trial: www.pluto.bham.ac.uk
NICE interventional procedure guideline: www.nice.org.uk/guidance/IPG202/?c=91520
Fetal movement charts Definition
A fetal movement chart is a record, usually kept by the mother, of her baby’s movements. The objective is to monitor the baby’s wellbeing, and to alert caregivers to ill-health or the risk of intrauterine death. The baby’s movements may also be monitored qualitatively, without recording on a chart. Epidemiology
Annual global stillbirths are estimated at 3.2 million (uncertainty range 2.5–4.1 million). Stillbirth rates range from 5 per 1000 in rich countries to 32 per 1000 in south Asia and sub-Saharan Africa. In South Africa, the commonest cause of perinatal loss is unexplained intrauterine death. Maternal perception of decreased fetal movements affects 5–15% of pregnancies. Pathology
Fetuses have remarkable mechanisms for conserving oxygen and energy when deprived, including preferential circulation to essential organs and reduced breathing and body movements. Movements may also be affected by central nervous system pathology, medication and reduced amniotic fluid volume. Betamethasone administration may suppress the diurnal increase in movements in the afternoon, but not affect the morning movement pattern. Aetiology
Risk factors for intrauterine death include maternal illness such as hypertensive disorders, diabetes mellitus, thyroid disease, and intrahepatic cholestasis; Chronic smoking, placental dysfunction with restricted fetal growth, placental abruption, amniotic fluid infection, congenital infections, fetal anaemia (from isoimmunization and other causes), fetomaternal haemorrhage, congenital anomalies and post maturity (>42 weeks for singletons, >38 weeks for twins). Prognosis
Reduced fetal movement has poor specificity for fetal ill-health. Thus the prognosis for babies with reduced movements is in general good.
The most important question regarding the use of fetal movement assessment (as for any screening test), is whether it is likely to do more good than harm. Potential harms include anxiety induced by the screening procedure, and a cascade of unnecessary interventions, particularly when the screening test has a high false-positive rate and the population being screened is at low risk. In the case of fetal movement assessment, the matter is made worse by the fact the ‘confirmatory’ test usually used, cardiotocography, also has a high false-positive rate.
The evidence of effectiveness of fetal movement monitoring is not straightforward. Two prospective cohort studies found a reduction in stillbirth when fetal movement counting was used, as did one quasi-randomized trial with allocation based on initial hospital booking number.
A systematic review found no randomized trials comparing fetal movement counting with no fetal movement counting. A large cluster randomized trial (>68 000 women) compared routine formal fetal movement counting with fetal movement counting at the discretion of the caregiver. The potential effect on perinatal outcome may have been masked by contamination of the ‘control’ group: the rate of antepartum late fetal deaths in both groups was considerably lower than it was prior to the commencement of the study. The potential effectiveness of routine over discretionary fetal movement counting is suggested by the fact that, when fetal movements were formally counted, there were more babies with subsequent unexplained late fetal deaths who were alive when first admitted to hospital (11/59 versus 6/58). However, the warnings did not translate into fewer deaths, mainly because of falsely reassuring fetal testing, mainly cardiotocography, and clinical error. There is thus indirect evidence that fetal movement counting may be effective in screening for babies at risk of intrauterine death, but to date no direct evidence of reduced perinatal mortality.
The recommendation of the UK NICE guidelines is that formal fetal movement counting should not be used routinely. Clinical approach to reduced fetal movements
Diagnosis
History
Fetal movements may be evaluated qualitatively by asking mothers to report their perception of reduced movements, or semiquantitatively by asking mothers to record movements on a chart. Various modifications of the ‘count to 10’ method developed in Cardiff, which measures the time taken for 10 movements to occur, have been found to enjoy better compliance than counting the number of movements over a specified time period. In low-risk Japanese women, the count to 10 time was almost the same from 22 weeks (10.9; 7.3–18.0 (median; interquartile range)) until 32 weeks (10.0; 6.2–15.6), then gradually increased toward 40 weeks (14.8; 9.5–24.0). Its 90th percentile was approximately 25 at 22–36 weeks and 35 minutes at 37–40 weeks.
Examination
The baby’s heart is auscultated to confirm that the baby is alive. Auscultation of a regular heart rate does not confirm the baby’s wellbeing. Examination of the mother is directed towards possible causes for reduced movements, and clinical assessment of the baby’s growth (such as by plotting serial measurements of the symphysis–fundus distance) and volume of amniotic fluid. Evaluation of the baby’s wellbeing may include clinical assessment of the baby’s movement or heart rate response to vibro-acoustic stimulation. The sound produced by an electronic vibro-acoustic stimulator may be mimicked by placing the base of an empty soft drink can against the mother’s abdomen, steadying it with a thumb and middle finger on the rim, depressing the ring opener with the index finger and allowing it to snap back.
Investigations
Various electronic methods of measuring fetal movements have been developed, but are not in routine clinical practice. Investigations are directed towards other methods of assessing the baby. If available, cardiotocography is often used to assess the baby’s wellbeing, although systematic review of cardiotocography for antepartum fetal assessment has shown an association with a trend to increased perinatal deaths. Vibro-acoustic stimulation but not manual manipulation has been shown to evoke fetal movements and thus facilitate fetal heart rate testing. Ultrasound is used to assess growth, amniotic fluid volume, anatomy, organ blood flow and umbilical artery resistance. Chronic placental hypoxia results in fetoplacental vasoconstriction with increased umbilical artery resistance index, a situation analogous to pulmonary hypertension in response to chronically hypoxic lungs. Observation of fetal breathing movements indicates that amniotic fluid infection is unlikely. The biophysical profile is a scoring system with 2 points each for adequate fetal body movement, tone, breathing, and amniotic fluid assessed by ultrasound, and reactive cardiotocography. There is to date inadequate evidence of effectiveness to support its use.
In some cases amniocentesis may be useful to exclude amniotic fluid infection and confirm fetal lung maturity.
Counselling
As for other methods of prenatal screening (and screening in general), even a simple recommendation to monitor the baby’s movements may generate anxiety.
The perception of reduced fetal movements is a cause of considerable anxiety, particularly in women with previous pregnancy losses. Careful explanation of the implications and limitations of the test is important.
Management
The management of reduced fetal movements must take into account several parameters:
• evidence of fetal compromise, and the strength of this evidence
• any identifiable causes for fetal compromise
• the gestational age of the baby.
Where the mother’s condition is contributing to fetal compromise and the baby is reasonably stable, the first priority is to optimize the mother’s condition.
Very rarely, the baby may have a condition amenable to intrauterine treatment, such as transfusion for anaemia or medical treatment for a tachyarrhythmia. Equally rarely, the baby’s condition may be considered irretrievable, for example severe central nervous system anomalies or extreme prematurity, and the parents may be counselled about the option of conservative care.
General measures to improve the baby’s condition include ensuring that the mother avoids the supine position, and tocolysis if the uterus in contracting.
In most cases the definitive management involves deciding on the optimal timing for delivery of the baby. Here the risks of intervention and prematurity need to be weighed against the risk of intrauterine deterioration or death if managed expectantly. If delivery is considered to be in the mother and baby’s best interest, the method of delivery will depend on the baby’s condition and the likelihood that the baby will tolerate labour induction (as opposed to Caesarean section).
Follow-up/recurrence/future pregnancy planning
When reduced fetal movements precedes a pregnancy loss, it is most important to counsel the parents about the implications of the loss. Most parents will want to know at least three things: What was the cause of the loss? Was it our fault? Will it happen again? To address these questions, considerable attention must be paid to ascertaining the cause of the loss, including clinical examination of the baby, genetic tests if indicated, tests for maternal endocrine disorders and infections, placental histology, and post-mortem examination of the baby. Further reading
de Heus R, Mulder EJ, Derks JB, et al. Differential effects of betamethasone on the fetus between morning and afternoon recordings. J Matern Fetal Neonatal Med 2008;21:549–54.
de Vries JI, Fong BF. Changes in fetal motility as a result of congenital disorders: an overview. Ultrasound Obstet Gynecol 2007;29:590–9.
Froen JF, Heazell AE, Tveit JV, et al. Fetal movement assessment. Semin Perinatol 2008;32:243–6.
Gibb D, Arulkumaran S. Fetal monitoring in practice, 2nd edn. Oxford: Butterworth Heinemann 1997.
Grivell RM, Wong L, Bhatia V. Regimens of fetal surveillance for impaired fetal growth (Protocol). Cochrane Database Syst Rev 2008;2:CD007113.
Habek D. Effects of smoking and fetal hypokinesia in early pregnancy. Arch Med Res 2007;38:864–7.
Heazell AE, Green M, Wright C, et al. Midwives’ and obstetricians’ knowledge and management of women presenting with decreased fetal movements. Acta Obstet Gynecol Scand 2008;87:331–9
Heazell AE, Froen JF. Methods of fetal movement counting and the detection of fetal compromise. J Obstet Gynaecol 2008;28:147–54.
Hofmeyr GJ, Neilson JP, Alfirevic Z, et al. A Cochrane pocketbook: pregnancy and childbirth. Chichester: Wiley 2008.
Kuwata T, Matsubara S, Ohkusa T, et al. Establishing a reference value for the frequency of fetal movements using modified ‘count to 10’ method. J Obstet Gynaecol Res 2008;34:318–23.
Lalor JG, Fawole B, Alfirevic Z, Devane D. Biophysical profile for fetal assessment in high risk pregnancies. Cochrane Database Syst Rev 2008;1:CD000038.
Mancuso A, De Vivo A, Fanara G, et al. Effects of antepartum electronic fetal monitoring on maternal emotional state. Acta Obstet Gynecol Scand 2008;87:184–9
Martin CB Jr. Normal fetal physiology and behavior, and adaptive responses with hypoxemia. Semin Perinatol 2008;32:239–42.
Neldam S. Fetal movements as an indicator of fetal well-being. Lancet 1980;1:1222–4.
Nishihara K, Horiuchi S, Eto H, Honda M. A long-term monitoring of fetal movement at home using a newly developed sensor: An introduction of maternal micro-arousals evoked by fetal movement during maternal sleep. Early Hum Dev 2008;84:595–603.
Neilson JP, Alfirevic Z. Doppler ultrasound for fetal assessment in high risk pregnancies. Cochrane Database Syst Rev 1996;4:CD000073.
N Pattison, L McCowan. Cardiotocography for antepartum fetal assessment. Cochrane Database Syst Rev 1999;1:CD001068.
Reddy UM. Prediction and prevention of recurrent stillbirth. Obstet Gynecol 2007;110:1151–64.
Sinha D, Sharma A, Nallaswamy V, et al. Obstetric outcome in women complaining of reduced fetal movements. J Obstet Gynaecol 2007;27:41–3.
Stanton C, Lawn JE, Rahman H, et al. Stillbirth rates: delivering estimates in 190 countries. Lancet 2006;367:1487–94.
Tan KH, Sabapathy A. Fetal manipulation for facilitating tests of fetal wellbeing. Cochrane Database Syst Rev 2001;4:CD003396. Tan KH, Smyth R. Fetal vibroacoustic stimulation for facilitation of tests of fetal wellbeing. Cochrane Database Syst Rev 2001;1:CD002963.
Troyano Luque JM, Maeda K, et al. Fetal extremity kinetics quantified with Doppler ultrasonography. J Perinat Med 2008;36:82–6.
Westgate J, Jamieson M. Stillbirths and fetal movements. NZ Med J 1986;99:114–16.
Mangesi L, Hofmeyr GJ. Fetal movement counting for assessment of fetal wellbeing. Cochrane Database Syst Rev 2007;1:CD004909. Internet resources
www.marchofdimes.com/professionals/14332_1198.asp Patient resources
Stillbirth and neonatal death society: www.uk-sands.org/
Fetal nuchal translucency Definition
Nuchal translucency (NT) is a black space seen by ultrasound behind the fetal neck and is produced by the collection of fluid under the skin.
• NT is observed in all fetuses between 11 and 13 weeks of pregnancy.
• In fetuses with trisomy 21 and other abnormalities, the size of NT tends to be higher than in normal fetuses. Epidemiology
The median and 95th centiles of fetal NT increase with fetal crown–rump length. In a population of 100 000 fetuses 200 have trisomy 21, another 200 have other chromosomal defects and 99 600 are euploid.
• In a few (5%) of euploid fetuses the NT is above the 95th centile. Therefore, 4800 euploid fetuses (5% of 99 600) would have high NT.
• In many (75%) fetuses with trisomy 21 and other chromosomal defects the NT is above the 95th centile. Therefore, 150 fetuses with trisomy 21 (75% of 200) and 150 fetuses with other chromosomal defects (75% of 200) have high NT.
• Therefore, even when the scan shows a high NT the majority of fetuses are euploid (4800 are euploid and 300 have chromosomal defects). Diagnosis
The diagnosis of increased fetal NT is made by ultrasound examination at 11–13 weeks of gestation.
The reasons for selecting 11 weeks as the earliest gestation are
• if after the screening test the parents choose to have CVS (chorionic villus sampling) this is not safe before 11 weeks because it can cause limb defects
• many major fetal abnormalities can be diagnosed at the NT scan, provided the minimum gestation is 11 weeks. With earlier scanning the abnormalities can be missed because the fetus is too small and the various organs are not developed enough to be visible.
The reasons for selecting 13 weeks and 6 days as the upper limit are
• to provide women with affected fetuses the option of first rather than second trimester termination
• even in fetuses with chromosomal abnormalities the high NT usually disappears after 13 weeks. Pathophysiology
Increased fetal NT is associated with a heterogeneous group of conditions, suggesting that there may not be a single underlying mechanism for the collection of fluid under the skin of the fetal neck.
Possible mechanisms include the following:
• Cardiac defects/dysfunction: in both chromosomally abnormal and euploid fetuses there is a high association between increased NT and cardiac defects.
• Venous congestion in the head and neck: this could result from constriction of the fetal body as encountered in amnion rupture sequence and superior mediastinal compression found in diaphragmatic hernia or the narrow chest in skeletal dysplasias.
• Altered composition of the extracellular matrix: many of the component proteins of the extracellular matrix are encoded on chromosomes 21, 18, or 13. Immunohistochemical studies, examining the skin of chromosomally abnormal fetuses, have demonstrated specific alterations of the extracellular matrix which may be attributed to gene dosage effects. Altered composition of the extracellular matrix may also be the underlying mechanism for increased fetal NT in an expanding number of genetic syndromes, which are associated with alterations in collagen metabolism (such as achondrogenesis type II), abnormalities of fibroblast growth factor receptors (such as achondroplasia), or disturbed metabolism of peroxisome biogenesis factor (such as Zellweger’s syndrome).
• Failure of lymphatic drainage: a possible mechanism for increased NT is dilatation of the jugular lymphatic sacs, because of developmental delay in the connection with the venous system, or a primary abnormal dilatation or proliferation of the lymphatic channels interfering with a normal flow between the lymphatic and venous systems. Immunohistochemical studies in nuchal skin tissue from fetuses with Turner syndrome have shown that the lymphatic vessels in the upper dermis are hypoplastic. In chromosomally normal fetuses with increased NT, deficient lymphatic drainage, due to hypoplastic or aplastic lymphatic vessels, is found in association with Noonan syndrome and congenital lymphoedema. In congenital neuromuscular disorders, such as fetal akinesia deformation sequence, myotonic dystrophy and spinal muscular atrophy, increased NT may be the consequence of impaired lymphatic drainage due to reduced fetal movements.
• Fetal anaemia: this is associated with a hyperdynamic circulation and fetal hydrops develops when the haemoglobin deficit is more than 7 g/dL. This is true for both immune and non-immune hydrops fetalis. However, in red blood cell isoimmunization severe fetal anaemia does not occur before 16 weeks of gestation, presumably because the fetal reticuloendothelial system is too immature to result in destruction of antibody coated erythrocytes. Consequently, red blood cell isoimmunization does not present with increased fetal NT. In contrast, genetic causes of fetal anaemia (α-thalassaemia, Blackfan–Diamond anaemia, congenital erythropoietic porphyria, dyserythropoietic anaemia, Fanconi anaemia) and possibly congenital infection-related anaemia can present with increased fetal NT.
• Fetal hypoproteinaemia: this is implicated in the pathophysiology of both immune and non-immune hydrops fetalis. In the first trimester, hypoproteinaemia due to proteinuria may be the underlying mechanism for the increased NT in fetuses with congenital nephrotic syndrome.
• Fetal infection: in about 10% of cases of ‘unexplained’ second- or third trimester fetal hydrops, there is evidence of recent maternal infection and, in these cases, the fetus is also infected. In contrast, in euploid pregnancies with increased NT, only 1.5% of the mothers have evidence of recent infection and the fetuses are rarely infected. Therefore, in pregnancies with increased NT the prevalence of maternal infection with the TORCH group of organisms may not be higher than in the general population. Increased NT in euploid fetuses need not stimulate the search for maternal infection unless the NT evolves into second or third trimester nuchal oedema or generalized hydrops. The only infection that has been reported in association with increased NT is parvovirus B19. In this condition the increased NT has been attributed to myocardial dysfunction or fetal anaemia due to suppression of haemopoiesis. Implications of increased NT
• Chromosomal defects: The prevalence of chromosomal defects increases exponentially with NT thickness from 0.2% for those with NT between the 5th and 95th centiles to 65% for NT of 6.5 mm or more. In the chromosomally abnormal group, about 50% have trisomy 21, 25% have trisomy 18 or 13, 10% have Turner’s syndrome, 5% have triploidy and 10% have other chromosomal defects.
• Fetal death: In chromosomally normal fetuses, the prevalence of fetal death increases with NT thickness from about 1% for those with NT between the 95th and 99th centiles to about 20% for NT of 6.5 mm or more. The majority of fetuses that die do so by 20 weeks and they usually show progression from increased NT to severe hydrops.
• Major fetal abnormalities: These are defined as those requiring medical and/or surgical treatment or conditions associated with mental handicap. The prevalence of major fetal abnormalities in chromosomally normal fetuses increases with NT thickness, from 1.5%, in those with NT below the 95th centile, to 2.5% for NT between the 95th and 99th centiles, and exponentially thereafter to about 45% for NT of 6.5 mm or more. Management
Although increased fetal NT thickness is associated with abnormalities and fetal death, the majority of babies survive and develop normally. After the diagnosis of increased NT the aim must be to distinguish as accurately and quickly as possible between those that are likely to have problems from those where the baby is likely to be normal.
Fetal NT between the 95th centile and 99th centile (3.5 mm)
The parents should be counselled of the possible increased risk for chromosomal defects. The decision by the parents in favour or against fetal karyotyping will depend on the patient-specific risk for chromosomal defects, which is derived from the combination of maternal age, sonographic findings and serum free β-human chorionic gonadotropin and PAPP-A.
A detailed scan should be carried out at 11–13 weeks and again at 20 weeks in search of major abnormalities. If no obvious abnormalities are seen the parents should be reassured that their baby is likely to be live born and develop normally. The chances that there would be any problems are not higher than in fetuses without increased NT.
Fetal NT above 3.5 mm
This is found in about 1% of pregnancies.
• The risk of chromosomal defects is very high and the first line of management of such pregnancies should be the offer CVS for fetal karyotyping. In patients with a family history of genetic syndromes that can be diagnosed by DNA analysis, the CVS sample can also be tested for these syndromes.
• A detailed scan should be carried out at 11–13 weeks in search of major abnormalities and genetic syndromes. A detailed scan is also carried out a couple of weeks later and again at 20 weeks.
• If no obvious abnormalities are seen and the NT has completely resolved, the parents should be reassured that their baby is likely to be live born and develop normally. The chances that the baby will have a serious abnormality or neurodevelopmental delay may not be higher than in the general population.
• If no obvious abnormalities are seen but there is persistence of increased NT at 14–16 weeks and evolution to nuchal oedema or hydrops fetalis at 20–22 weeks, it is possible that there is congenital infection or a genetic syndrome. Maternal blood should be tested for toxoplasmosis, cytomegalovirus, and parvovirus B19. Follow-up scans should be carried out every 4 weeks to define the evolution of the oedema. Consideration should be given to DNA testing for certain genetic conditions, such as Noonan syndrome, even if there is no family history for these conditions. The parents should be counselled that there is a 10% risk of perinatal death or a live birth with a genetic syndrome that could not be diagnosed prenatally. The risk of neurodevelopmental delay in the survivors is 3–5%. Further reading
Nicolaides KH. The 11-13 + 6 week scan. Fetal Medicine Foundation, London, 2004. Available in pdf at www.fetalmedicine.com/ Internet resources
www.fetalmedicine.com/ Fetal abnormalities: hydrops Definition
Hydrops is defined as an abnormal accumulation of serous fluid in at least two fetal compartments, including ascites, pleural or pericardial effusions, and skin oedema (Fig.7.17.1). Epidemiology
Hydrops is a rare finding, affecting about 1 in 2000 pregnancies. As it can be caused by a number of recessive genetic disorders, it is more common in countries where those disorders are common. Thus, hydrops may be as common as 1 in 500 due to homozygous α-thalassaemia in South-East Asia, or glucose-6-phosphate dehydrogenase deficiency in Mediterranean countries. Aetiology
In general terms, hydrops can be due to cardiac failure, obstructed lymphatic flow, or decreased plasma osmotic pressure.
Multiple pregnancy
The finding of hydrops in one fetus of a monochorionic pair is typical of severe twin–twin transfusion or twin reversed arterial perfusion syndromes. This is due to overload and cardiac failure of the recipient twin.
Fetal anaemia
Severe anaemia causes cardiac failure and the finding of hydrops. Hydrops will usually only result from severe anaemia, i.e. when the fetal haemoglobin deficit is more than 7 g/dL below the normal mean for gestation.
• Red cell alloimmunization: until the advent of anti-D prophylaxis, the most common cause of hydrops fetalis was blood group isoimmunization. Although usually due to Rhesus blood group antigens, other blood group antigens including C, E, and Kell antigens can cause fetal haemolytic anaemia
• parvovirus B19 infection
• homozygous α-thalassaemia
• glucose-6-phosphate dehydrogenase deficiency (X-linked, dominant)
• glucose phosphate isomerase deficiency (autosomal recessive)
• pyruvate kinase deficiency (autosomal recessive)
• Diamond–Blackfan syndrome (autosomal dominant)
• fetomaternal haemorrhage
• intrafetal haemorrhage, e.g. intracranial, into a large ovarian cyst.
Structural abnormalities of the fetus or placenta
The most common causes for hydrops in this group are due to cardiac failure due to congenital heart defects and arrhythmias. Thoracic abnormalities can cause cardiac compression, again leading to cardiac failure.
Cardiac abnormalities
• About 40% of cases in this group are due to structural cardiac defects, including left or right hypoplastic heart, large atrioventricular septal defects, or endocardial fibroelastosis.
• Cardiac arrhythmias: both tachy- and bradyarrhythmias. Causes include supraventricular tachycardia and complete heart block due to maternal anti-Ro or anti-La antibodies. Absence of a natural pacemaker (sino-atrial node) can occur in isomerism.
• Cardiomyopathies and cardiac rhabdomyoma.
Thoracic abnormalities
• Cystic lung lesions (congenital cystic adenomatoid malformation, pulmonary sequestration), or other intrathoracic neoplasms.
• Congenital diaphragmatic hernia.
• Congenital high airway obstruction syndrome (CHAOS).
Arteriovenous shunts
High output cardiac failure can be secondary to abnormal arteriovenous communications. These include:
• placental chorioangiomas
• fetal sacrococcygeal teratomas
• other placental of fetal tumours.
Fetal syndromes
• Chromosomal: most commonly trisomies 21, 18, 13, Turner’s syndrome or triploidy.
• Genetic: A large number of genetic diseases can present with hydrops fetalis. The more common causes are Noonan syndrome, glycogen storage diseases, lysosome storage diseases, fetal akinesia/deformation sequence, achondrogenesis, Fraser syndrome, multiple pterygium syndrome and Smith–Lemli–Opitz syndrome, tuberous sclerosis (autosomal dominant).

Fig. 7.17.1 Ultrasound diagnosis of (a) skin oedema and (b) fetal ascites in fetal hydrops.
Fetal congenital infection
These are most commonly due to early pregnancy congenital infection with cytomegalovirus or parvovirus B19 (see Anaemia, above). Other causative organisms are toxoplasmosis, coxsackie virus, listeria and syphilis. Prognosis
The most important determinants are the underlying cause and the gestation at presentation. Estimates of mortality vary widely, but most case series report 60–90% mortality. Many cases are due to underlying congenital malformations, genetic or chromosomal abnormalities which in themselves are fatal.
Clinical approach: diagnosis
Referral to a tertiary level fetal medicine unit is indicated. This is essential not only for invasive testing to be performed, but will also allow the woman to benefit from multidisciplinary team approach. This will usually include a fetal medicine expert, cardiologist, geneticist, and other allied specialists.
History
• Previous pregnancy including previously affected pregnancies
• previous fetal congenital abnormalities or fetal death
• previous fetal or neonatal anaemia or jaundice
• previous history of rhesus or other blood group antibodies
• drug history, including recreational drug use
• haemoglobinopathy
• recent of illness or flu-like symptoms
• personal or family history of genetic or metabolic diseases.
Examination/ultrasound findings
• In monochorionic twin pregnancy the finding of hydrops is usually associated with severe twin–twin transfusion syndrome (TTTS). The hydropic twin will have polyhydramnios, a dilated heart with reduced myocardial contractility and ductus venosus Doppler will usually be abnormal. The diagnosis of TTTS is confirmed if the donor co-twin exhibits growth restriction, oligo- or anhydramnios (stuck twin), an unfilled bladder, or abnormal umbilical artery Doppler.
• Doppler measurement of the peak systolic velocity (PSV) in the middle cerebral artery (MCA) is a useful tool in predicting a hyperdynamic fetal circulation. An increased PSV is suggestive of a hyperdynamic circulation, and this is most commonly due to fetal anaemia, which can be caused by alloimmunization, parvovirus infection, congenital fetal anaemia, or fetomaternal haemorrhage. It can also be present due to a fetal or placental tumour or arteriovenous malformation.
• A detailed ultrasound examination looking for possible abnormalities outlined above, including specialist fetal echocardiography should be performed.
Investigations
These depend on the history and detailed ultrasound examination findings.
Maternal
• Blood type/blood group
• CMV, toxoplasma, and parvovirus titres
• If fetal bradyarrhythmia is suspected an antibody screen (Anti-Ro, Anti-La) should be performed
• Kleihauer test if fetomaternal haemorrhage is suspected.
Fetal
Invasive testing should be discussed with the parents. If the hydrops was diagnosed at the 11–14-week scan, chorionic villus sampling will allow fetal karyotype analysis and storage of fetal DNA. Fetal blood sampling should be considered if possible, as this allows measurement of other fetal parameters.
• Fetal karyotype.
• Full blood count.
• Coombs test.
• Metabolic tests may be indicated depending on family history.
• Serum protein may be indicated if hypoproteinaemia is suspected.
• Fetal DNA should always be stored for potential future analysis of as yet unrecognized genetic diseases.
Counselling and management
From the point of view of counselling, hydrops could be classified into treatable and untreatable causes. Treatable causes include.
Twin–twin transfusion syndrome
Left untreated, the mortality of this condition exceeds 90%, with significant neurological morbidity in 30–50% of surviving twins. Serial amnioreduction and fetoscopic laser ablation of the placental vascular anastomoses have been used for treatment of the condition. There is a significant increase in survival rates and reduction in neurological morbidity with the use of laser ablation compared with amnioreduction in severe TTTS. Overall survival rates after laser therapy range from 56% to 62%, survival of at least one twin was reported in 76–83% of cases, and neurological morbidity in about 5–10% of survivors.
Twin reversed arterial perfusion syndrome
Fetoscopic laser coagulation was assessed in a multicentre study and resulted in an overall survival rate of the pump twin of 80%. Preterm premature rupture of the membranes prior to 34 weeks’ gestation occurred in 18%.
Fetal anaemia
• The procedure related fetal loss rate after fetal blood transfusion is approximately 2–3% in expert hands.
• Parvovirus infection: this has a generally good outcome if treated appropriately with fetal blood transfusion. Generally, only one or two transfusions may be necessary. One report has suggested that there is an increased incidence of neurodevelopmental delay in children born from pregnancies affected with Parvovirus infection treated with transfusion. In this series five of 16 infants (32%) exhibited some delayed psychomotor development at the age of 6–8 years. Previous reports however did not find such an association.
• Fetal anaemia secondary to immunisation: fetal blood will continue to be destroyed by maternal antibodies. The number of transfusions will depend on the gestational age at onset of the disease, the degree of anaemia, and the rate of red cell breakdown. Repeat transfusions will be necessary in the majority of cases. After 34 weeks of gestation most practitioners would suggest delivery, as neonatal outcome is likely to be good and ex utero treatment removes the procedure related risks of intrauterine transfusion.
Cardiac arrhythmias
Treatment of fetal cardiac arrhythmias has been reported. Tachyarrhythmias may respond to maternal administration of flecainide, amiodarone, or sotalol.
Chorioangiomas and other AV malformations
Successful interstitial laser therapy of placental chorioangiomas and other AV malformations have been reported. However, due to the rare nature of these conditions it is difficult to assess success rates. Such treatment is warranted in cases of severe progressive hydrops where the prognosis for the pregnancy is poor.
Non-treatable causes of hydrops
When fetal hydrops remains unexplained the prognosis for the pregnancy is very poor. In the presence of an underlying fetal structural abnormality, counselling and management will depend on the precise findings. Options for the pregnancy include monitoring, and elective pregnancy termination. Further reading
Dembinski J, Haverkamp F, Maara H, et al. Neurodevelopmental outcome after intrauterine red cell transfusion for parvovirus B19-induced fetal hydrops. Br J Obstet Gynaecol 2002;109:1232–4.
Fox CD, Kilby, Khan K.S. Contemporary treatments for twin–twin transfusion syndrome. Obstet Gynecol 2005;105:1469–77.
Hecher K, Lewi L, Gratacos E, et al. Twin reversed arterial per-fusion: fetoscopic laser coagulation of placental anastomoses or the umbilical cord. Ultrasound Obstet Gynecol. 2006;28:688–91.
Heegaard ED, Brown KE. Human parvovirus B19. Clin Microbiol Rev 2002;15:485–505.
Machin GA. Hydrops revisited: literature review of 1, 414 cases published in the 1980s. Am J Med Genet 1989;34:366–90.
Nagel HT, de Haan TR, Vandenbussche FP, et al. Long-term outcome after fetal transfusion for hydrops associated with parvovirus B19 infection. Obstet Gynecol 2007;109:42–7.
Nicolaides KH, Fontanarosa M, Gabbe SG, Rodeck CH. Failure of ultrasonographic parameters to predict the severity of fetal anemia in rhesus isoimmunization. Am J Obstet Gynecol 1988;158:920–6.
Simpson JM. Fetal arrhythmias. Ultrasound Obstet Gynecol 2006;27:599–606.
Van Kamp IL, Klumper FJ, Oepkes D, et al. Complications of intrauterine intravascular transfusion for fetal anemia due to maternal red-cell alloimmunization. Am J Obstet Gynecol 2005;192:171–7. Useful websites
Hydrops fetalis: www.emedicine.com/ped/topic1042.htm Parvovirus: www.hpa.org.uk/infections/topics_az/parvovirus/gen_info.htm Patient resources
Hydrops: www.patient.co.uk/showdoc/40024818/
Rhesus disease: www.patient.co.uk/showdoc/40000464/
Invasive procedures Prenatal diagnosis: chorionic villus sampling and amniocentesis
It is estimated that 5% of the pregnant population (30 000 women a year in the UK) are offered invasive prenatal diagnosis (RCOG), in the majority of cases this involves a choice between chorionic villous sampling (CVS) or amniocentesis.
Indications
The most common indication is for performance of fetal chromosome analysis, usually due to screen positive screening test. Other indications include analysis for genetic disease in susceptible pregnancies (for example where both parents are known to be carriers of a recessive genetic disease, or a previous history of a genetic problem).
Procedure outline
CVS is performed from 10 to 14 weeks. With transabdominal CVS a fine needle is introduced through the mother’s lower abdomen into the placenta under ultrasound guidance. Transcervical CVS is used much less commonly.
Amniocentesis is performed from 15 weeks onwards. A fine needle is introduced through the mother’s lower abdomen or amniotic sac under ultrasound guidance. Between 10 and 20 mL of amniotic fluid is aspirated and this contains fetal cells (from skin, lungs, and urinary tract).
Efficacy
With either technique cells are analysed for chromosomal abnormalities and DNA can be extracted for molecular analysis.
One advantage of CVS is that it is performed much earlier in gestation (between 10 and 14 weeks). As abnormalities are identified at an earlier stage, pregnancy termination may be easier, performed surgically rather than medically, and therefore may be more acceptable. A disadvantage is a small risk of confined placental mosaicism that may result in inconclusive results making additional invasive tests necessary.
Safety
Historically CVS has been thought to have higher procedure-related loss than amniocentesis (Table 7.18.1). However, evidence from meta-analysis and recent series suggest that loss rates after transabdominal CVS are similar to amniocentesis in specialized centres. This may be due to increased experience compared with the earlier studies (operator experience has been shown to have a major impact on procedure-related loss rates), taking account of the earlier gestation at which CVS is performed, and a general move towards a transabdominal approach (a transcervical approach has been associated with a higher CVS loss rate).
Table 7.18.1 Comparison between CVS and amniocentesis

In utero therapeutic interventions
Fetal blood sampling and in utero transfusion
Fetal blood sampling (FBS) is also known as cordocentesis or percutaneous umbilical blood sampling (PUBS).
Indications
Suspected fetal anaemia or thrombocytopenia. Given the speed of results from CVS and amniocentesis, cordocentesis is rarely used for fetal chromosome analysis.
Middle cerebral artery velocity by Doppler scanning (MCA-PSV) is now the preferred method for detection of fetal anaemia and avoids the need for invasive amniocentesis or repeat fetal blood sampling. A threshold value (usually 1.5 MoM) is used to determine when FBS with possible in utero transfusion (IUT) should be performed.
Transfusion may also be needed in cases of haemolytic anaemia due to non-Rhesus antigens, non-haemolytic anaemia (e.g. fetal parvovirus infection) and fetal alloimmune thrombocytopenia when intrauterine platelet transfusions are required.
Procedure outline
A needle is introduced into the umbilical vein under ultrasound guidance. A sample of fetal blood is rapidly analysed for haematocrit and also sent to haematology. Cross-matched, irradiated, and CMV-negative blood or compatible platelet hyperconcentrates are then transfused as necessary. Rapid delivery in the event of severe fetal distress may be necessary assuming fetal viability.
Efficacy
Modern management of Rhesus disease with MCA-PSV monitoring and judicious use of FBS and IUT has successfully reduced the perinatal mortality associated with this previously devastating condition. In utero transfusion in other cases of fetal anaemia can reverse related hydrops and reduce mortality.
Weekly in utero transfusion of platelets has been shown to be effective in preventing intracranial haemorrhage in severe cases of alloimmune thrombocytopenia, although there are cumulative loss rates of up to 10% for each pregnancy as a result.
Safety
There is a fetal loss of at least 1% for each procedure. These risks include fetomaternal haemorrhage, placental abruption, acute fetal distress (bradycardia), and chorioamnionitis with maternal sepsis. These have to be balanced against the risk of preterm delivery, and in most cases delivery, rather than continuing transfusions, is undertaken beyond 34 weeks. Laser ablation for twin–twin transfusion syndrome
Indication
Laser ablation is now the accepted best treatment for severe twin–twin transfusion syndrome (TTTS). The role of laser ablation in early TTTS is more controversial.
Outline of procedure
The aim is to divide the abnormal communicating vessels between monchorionic twins and prevent abnormal shunting of blood from the donor to recipient twin. The procedure is usually performed under regional or local anaesthesia under ultrasound guidance. A fetoscope is inserted into the amniotic sac of the recipient twin and anastomosing placental vessels are ablated using a laser.
After completion of surgery amnio drainage of the recipient twin is performed.
Efficacy
Fetoscopic laser, compared with serial amnioreduction or septostomy, both improves perinatal survival and reduces long-term neurological sequelae in survivors. A review of two randomized controlled trials showed less death of both infants per pregnancy (relative risk (RR) 0.49), less perinatal death (RR 0.59) and less neonatal death (RR 0.29) than in pregnancies treated with amnioreduction. More babies were alive without neurological abnormality at the age of six months in the laser group than the amnioreduction groups (RR 1.66) although this difference did not persist beyond six months of age. 1
In utero shunt insertion
Fetal shunts have been inserted in utero for drainage of obstructed lower urinary tracts and pleural effusions.
Outline of shunt insertion
A cannula or trocar is inserted transabdominally into the amniotic cavity under maternal local anaesthesia. This is guided through the fetal abdominal or chest wall into the bladder or pleural effusion. The entire procedure is performed under ultrasound guidance. A pigtail catheter is inserted through the cannula and positioned with one end in the target organ and the other draining into the amniotic cavity. The cannula is removed and the final position of the catheter confirmed with ultrasound
Fetal vesico-amniotic shunt
The evidence supporting shunt insertion is at present uncertain.
Indication
Fetal lower urinary tract outflow obstruction. The purpose of the shunt is to decompress the obstructed bladder and restore normal amniotic fluid dynamics with the aim of preventing the sequelae of renal dysplasia and pulmonary hypoplasia.
Efficacy
A randomized controlled trial is currently in progress (PLUTO) comparing shunt insertion with no shunt. A meta-analysis of three controlled trials comparing outcomes following shunt insertion (n = 59) with no insertion (n = 33) demonstrated an improvement in perinatal survival (OR 2.53). Overall survival rates after shunt insertion are still poor at 40%, reflecting the underlying poor prognosis. There is limited information as to the effect of shunt insertion on longer term outcomes such as need for dialysis (~25%) and long-term bladder and respiratory function.
Safety
The commonest complication is shunt displacement or blockage, occurring in 20–30% of cases, requiring a second shunt insertion. Premature labour and rupture of membranes, fetal trauma, and urinary ascites are all documented complications. Potential maternal complications include trauma to organs and infection though from the limited case series data these would appear to be rare. Pleuro-amniotic shunt
Indication
Large pleural effusions can lead to lung compression, hypoplasia and hydrops. The aim of shunt insertion is to reverse these. Case selection is difficult as there is uncertainty about the natural history of pleural effusions, and some will resolve spontaneously. This makes it difficult to balance the risks of treatment against the natural progression of the effusion.
Efficacy
Depending on the selection of cases, pleuro-amniotic shunts are effective at draining pleural effusions and allowing lung expansion (98%) and produce resolution of hydrops in 50% of cases. Treatment outcomes vary widely across case series and probably reflect differences in underlying aetiology and case selection.
Safety
An estimated 10% of fetuses will die as a result of shunt insertion; however, in the presence of hydrops there is a high background fetal loss rate. Displacement and blockage of the shunt occur in 20–30% of cases, traumatic haemothorax has also been reported. Balloon valvuloplasty
Indication
Severe aortic stenosis or pulmonary atresia with an intact ventricular septum. The aim is to prevent progressive damage to the ventricular muscle in utero, the development of hydrops and subsequent fetal death.
Careful case selection is needed given the uncertainty as to the safety and efficacy of valvuloplasty.
Procedure
Fetal positioning is critical for success. The procedure is conducted at 21–32 weeks’ gestation with maternal local anaesthesia and sedation under ultrasound guidance. A needle is passed through the fetal chest into the left or right ventricle and a guidewire passed across the aortic or pulmonary valve. A balloon catheter is inserted and inflated to dilate the stenotic valve.
Efficacy
There are very limited data with the largest case series of aortic valvuloplasty including 20 fetuses. Technical success was achieved in 70% of cases with subsequent growth of the aortic and mitral valves. The data are even more limited on pulmonary valvuloplasty with only 10 cases described in the literature.
Safety
Again data are extremely limited, but fetal bradycardia, pericardial effusion, balloon rupture and fetal death are all potential complications.
An intention-to-treat registry has been developed by the Association for European Paediatric Cardiology (www.aepc.org), which it is hoped will provide much needed evidence in the future. Further reading
Interventional procedures guidance: www.nice.org.uk/guidance
Wapner RJ. Invasive prenatal diagnostic techniques. Semin Perinatol 2005;29:401–4.
Brun JL, Mangione R, Gangbo, et al. Feasibility, accuracy and safety of chorionic villus sampling: a report of 10 741 cases. Prenat Diagn 2003;23:295–301.
Horger EO, et al. A single physician’s experience with four thousand six hundred genetic amniocenteses AmJOG 2001: 185: 279–88.
Mujezinovic F, Alfirevic Z. Procedure-related complications of amniocentesis and chorionic villous sampling: a systematic review. Obstet Gynecol 2007;110:687–94.
Fergus S, et al. The loss rates for invasive prenatal testing in a specialised obstetric ultrasound practice Aust NZ J Obstet Gynaecol 2002;42:1: 61.
Roberts et al. Cochrane Library 2008.
Tabor et al. Randomised controlled trial of genetic amniocentesis in 4606 low risk women. Lancet 1986; i: 1287–93. Patient resources
ARC (Antenatal Results and Choices): www.arc-uk.org
Intrauterine growth restriction (IUGR) Definition
IUGR or fetal growth restriction (FGR) is slowing of expected in utero growth causing the fetus not to reach its genetic growth potential. This should be differentiated from small for gestational age (SGA) babies whose estimated or birth-weight is below the 10th percentile for gestational age. Epidemiology
The frequency of SGA depends on the definition used; by definition it will occur in 10% of the population if it is defined as size below the 10th percentile, 5% if defined as below the 5th percentile, etc.
The frequency of FGR is much more difficult to assess, as this also depends on the exact definition used. Owing to the wide biological variability in fetal and newborn size and uncertainties in dating/gestation, identifying a fetus that is not growing at its genetic potential is difficult. Most estimates suggest that it occurs in around 3% of pregnancies.
FGR is an important condition as it is associated with increased perinatal morbidity and mortality as well as longer term adverse health outcomes, including cardiovascular disease and metabolic syndrome. Aetiology
In broad terms the causes are classified below.
Wrong dates
In order to plot fetal size accurately, gestational age must be known accurately. As last menstrual period dates (LMP) are either unknown or erroneous in around one-third of pregnancies the pregnancy should be dated by the first available ultrasound scan, preferably by measurement of the fetal crown–rump length (CRL) between 8 and 12 weeks of gestation. Dating in the second and third trimester is much less accurate and serial scans to assess fetal growth patterns may be needed.
Constitutionally small fetus
Estimated fetal weight (EFW) and birthweight centiles are used most commonly to diagnose FGR. This has the disadvantage of classifying a large proportion of constitutionally ‘normal small’ babies as being at risk. Careful examination of the fetus to differentiate between this and FGR is necessary to adequately assess risk, and this will usually involve careful assessment for abnormalities, Doppler assessment of the uteroplacental and fetoplacental circulation and serial growth assessment. It should be noted that data suggest that even when infants have SGA that is thought to be constitutional, there is an excess risk of perinatal and longer term morbidity.
Multiple pregnancy
There is an effect of fetal number on birthweight with increasing numbers of fetuses in higher order multiples being smaller at all gestations after around 24 weeks. These differences are presumed to be due to mechanical constraints of space or placental implantation, but other causes of FGR can of course affect multiple pregnancies.
Fetal abnormalities
There are a number of fetal conditions that can lead to FGR, and these can be inherent (such as a chromosomal abnormality) or acquired in utero (such as a congenital infection). The conditions will almost always have coexisting fetal abnormalities, and careful ultrasound examination should be mandatory in cases of severe FGR to assess for these. They include:
• fetal chromosomal abnormalities (particularly trisomy 18 and 13, and triploidy)
• rare genetic disorders (such as Silver Russell syndrome, Seckel’s syndrome)
• fetal congenital infections (including cytomegalovirus and toxoplasma and, more rarely, rubella and fetal varicella syndrome)
• abnormalities of the fetal circulation, such as single umbilical artery or abnormal course of the ductus venosus.
Placental insufficiency
Once wrong dates and constitutional smallness are excluded, this is the commonest cause. The condition overlaps with pre-eclampsia and placental abruption in its pathophysiology, which has its origins in the first trimester of pregnancy—there is evidence of altered uterine artery blood flow and markers of abnormal angiogenesis at 11–13 weeks. Placental insufficiency may also be ‘acquired’ by heavy maternal smoking or drug abuse (especially cocaine abuse), which may affect growth through abnormal placental function. Fetuses with FGR due to placental insufficiency are often described as having ‘asymmetrical’ growth restriction, with the abdominal circumference falling away from the normal range before the femur length, although head measurements are generally spared. Amniotic fluid is usually reduced, uterine artery Doppler usually shows high impedance to flow and umbilical artery blood flow may be reduced, absent or reversed. The condition is progressive and management is mainly aimed at balancing the risks of intrauterine hypoxia with those of indicated preterm delivery.
Placental chorioangiomas can also be a rare cause of FGR, although in these cases a discrete placental anatomical abnormality is usually visible. Prognosis
This depends mainly on the underlying cause and the gestational age and birthweight at delivery. When due to placental insufficiency, early-onset FGR (defined in most studies as leading to delivery before 34 weeks of gestation) has survival rates that are significantly lower than appropriately grown counterparts. There has to be a careful balance between early delivery (which carries a higher rate of neonatal complications) versus expectant management (with risks of inadvertent stillbirth or sequelae of intrauterine hypoxia). Each day in utero increases survival and intact survival by 1–2% and this is particularly evident at lower gestational ages. Clinical approach: diagnosis
Referral to a fetal medicine unit with appropriate expertise in the diagnosis and management of FGR is indicated.
History
• Assess pregnancy dating. If previous dating was accurate redating of the pregnancy must not be carried out at this late gestation.
• Previous pregnancy including previous pregnancies affected by SGA, FGR, pre-eclampsia or abruption.
• Previous or family history of genetic conditions or fetal congenital abnormalities.
• Drug history, including recreational drug use.
• Recent of illness or flu-like symptoms.
Examination/ultrasound findings
• Carefully measure fetal biometry and plot in relation to previous ultrasound scans.
• Assess fetal and placental anatomy, uterine, umbilical and middle cerebral artery Doppler, amniotic fluid, and fetal biophysical profile.
• If fetal growth velocity is maintained along a centile line, with normal fetal and placental anatomy, and normal Doppler, amniotic fluid, and fetal biophysical profile, wrong dates or constitutional smallness is the most likely cause.
• In fetal conditions, Doppler, amniotic fluid, and fetal behaviour may be normal or abnormal; increased amniotic fluid in the presence of severe early-onset FGR is an ominous sign. Fetal abnormalities suggestive of the causes outlined in aetiology, above, are usually present. If uterine and fetal Dopplers are normal despite early-onset severe FGR, a fetal condition is likely. Nevertheless, abnormal Dopplers may coexist in certain fetal abnormalities (e.g. triploidy). Targeted investigations for fetal chromosomal abnormalities or congenital infections should be carried out depending on the findings.
• When the cause for the FGR is placental insufficiency the majority of pregnancies will have Doppler abnormalities in the uterine and umbilical artery. On the other hand, late-onset FGR often has few Doppler abnormalities, such as an isolated reduction in impedance to flow through the middle cerebral artery despite normal umbilical artery Doppler. Fetal heart rate abnormalities may be present. Clinical Approach: management
Management should be targeted at the underlying cause. In early-onset FGR due to placental insufficiency there is a well-described progression of arterial to venous Doppler abnormalities which usually precedes abnormalities in biophysical profile and fetal heart rate. The sequential changes are a decrease in umbilical artery end diastolic flow (this the progresses through absent to reversed end diastolic flow); evidence of ‘brain sparing’, with a fall in the middle cerebral artery indices, followed by increasing ductus venosus Doppler indices. As outlined above, in late-onset FGR Doppler changes do not typically follow this pattern. A more detailed discussion of fetal monitoring can be found in the Chapters 7.3, Doppler ultrasound, and Chapter 7.1, Biophysical profile. Further reading
Barker DJ. Fetal growth and adult disease. Br J Obstet Gynaecol 1992;99:275–76.
Baschat AA, Cosmi E, Bilardo CM, et al. Predictors of neonatal outcome in early-onset placental dysfunction. Obstet Gynecol 2007;109:253–61.
Bernstein IM, Horbar JD, Badger GJ, et al: Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction: The Vermont Oxford Network. Am J Obstet Gynecol 2000;182:198–206.
Bilardo CM, Wolf H, Stigter RH, et al. Relationship between monitoring parameters and perinatal outcome in severe, early intrauterine growth restriction. Ultrasound Obstet Gynecol 2004;23:119–25.
Cosmi E, Ambrosini G, D’Antona D, et al. Doppler, cardiotocography, and biophysical profile changes in growth-restricted fetuses. Obstet Gynecol 2005;106: 1240–5.
Eixarch E, Meler E, Iraola A, et al. Neurodevelopmental outcome in 2-year-old infants who were small-for-gestational age term fetuses with cerebral blood flow redistribution. Ultrasound Obstet Gynecol 2008;32:894–9.
Hecher K, Bilardo CM, Stigter RH, et al. Monitoring of fetuses with intrauterine growth restriction: a longitudinal study. Ultrasound Obstet Gynecol 2001;18:564–70.
Manning FA. Antepartum fetal testing: a critical appraisal. Curr Opin Obstet Gynecol 2009;21:348–52.
Nyberg DA, Abuhamad A, Ville Y. Ultrasound assessment of abnormal fetal growth. Sem Perinatol 2004;28:3–22.
Thornton JG, Hornbuckle J, Vail A, et al. GRIT study group. Infant wellbeing at 2 years of age in the Growth Restriction Intervention Trial (GRIT): multicentred randomised controlled trial. Lancet 2004;364:513–20.
Turan OM, Turan S, Gungor S, et al. Progression of Doppler abnormalities in intrauterine growth restriction. Ultrasound Obstet Gynecol; 2008: 32: 160–7.
Turan S, Miller J. Baschat AA. Integrated testing and management in fetal growth restriction. Semin Perinatol 2008;32:194–200.
Vintzileos AM, Fleming AD, Scorza WE, et al. Relationship between fetal biophysical activities and umbilical cord blood gas values. Am J Obstet Gynecol 1991;165:707–13. Patient resources
www.childgrowthfoundation.org/ Multiple pregnancy Definition
This occurs when two or more ova are fertilized to form dizygotic (non-identical) twins or a single fertilized egg divides to form monozygotic (identical) twins. Chorionicity
Chorionicity refers to the number of placentas in a multiple pregnancy. Dizygotic twin pregnancies always have two placentas (dichorionic). There is one placenta for each fetus and they may be fused or separate. In monozygotic multiple pregnancies, the number of placentas depends on the timing ovum division. If the embryo splits before day 4 there are two placentas. Embryos that split late (days 4–12) will have a single placenta (monochorionic). Monochorionic twins are prone to significantly more pregnancy complications than dichorionic twins. Epidemiology
The incidence of twins is 1 in 80 and triplets 1 in 900 pregnancies. The use of assisted reproductive techniques has greatly increased the incidence of multiple pregnancies. Twinning is also slightly more common with previous multiple pregnancy, family history, increasing maternal age, and West African racial origin. Presentation
Early symptoms include exacerbation of the usual pregnancy-related symptoms (especially hyperemesis) and a uterus that is clinically large for dates. It is possible to confirm multiple pregnancies and its chorionicity in the first trimester by ultrasound. Complications
Vanishing twin
In a small, but significant proportion of pregnancies, one twin dies and is reabsorbed in the first trimester of pregnancy. Under these circumstances, serum screening for the risk of trisomy 21 may be unreliable.
Twin–twin transfusion syndrome
In monochorionic twin pregnancies, there can be unequal vascular sharing of the placenta with intertwin transfusion, leading to one twin developing growth restriction and the other cardiac failure.
Fetal growth restriction
Twins are usually smaller at any given gestational age than singletons. However, both mono- and dichorionic twins have a high prevalence of fetal growth restriction. The severity of the growth restriction and pregnancy outcome is often worse in monochorionic twins.
Premature delivery
The mean gestation of delivery is 37 weeks for twins and 31 weeks triplets. Multiple pregnancies have a higher incidence of preterm and severe preterm delivery.
Other complications
All maternal (anaemia, pre-eclampsia, placenta praevia) and fetal complications (cerebral palsy, perinatal mortality) of pregnancy are more common in multiple pregnancy except for prolonged pregnancy and fetal macrosomia. Management
Prenatal screening
Fetal abnormality is more common in multiple than singleton pregnancy, as a consequence of the effects of egg splitting (monozygotic) and the number of fetuses (dizygotic).
Nuchal translucency assessment at 10–14 weeks may used to screen for risk of aneuploidy. However, invasive prenatal diagnosis is complicated by the issue of zygosity, possible sample contamination, and the ethical dilemma of managing a twin pregnancy with one abnormal fetus. Under the latter circumstances, the parents may opt for selective termination, where the miscarriage rate for the co-twin is as high as 5–8%.
Antenatal
As most pregnancy complications are seen more frequently in twin pregnancy, they should be managed by a specialist with more frequent monitoring. Ultrasound scans should be undertaken to determine chorionicity (Fig. 7.20.1), fetal growth and signs of twin–twin transfusion syndrome (TTTS).
Fetal growth restriction is managed by regular monitoring, steroids for lung maturation, and early delivery for deteriorating Doppler blood flow indices. With severe TTTS, the appropriate management is fetoscopic laser surgery to divide the monochorionic intertwin vascular placental anastomoses.
Twin pregnancies with a short cervix (< 20 mm) are at significantly higher risk of preterm delivery. Cervical sutures and antibiotic prophylaxis have not shown to be of value and, in fact, may worsen pregnancy prognosis. The use of progesterone to reduce risk of preterm delivery appears promising and is being investigated currently.
Other pregnancy complications should be screened for and managed as for singleton pregnancy. Delivery
In dichorionic twins, if the first twin presents as cephalic, vaginal delivery may be attempted. Elective Caesarean section is indicated for the same reasons as in singleton pregnancy. Monochorionic twins and triplets are usually delivered by Caesarean section because of the risks of acute intertwin transfusion in the former, and high rate of emergency Caesarean and difficulty in fetal monitoring in the latter.

Fig. 7.20.1 The -sign of a dichorionic placenta.
Intrapartum
The main fetal risk in labour is thought to be as a result of hypoxia to the second twin during a protracted second stage and premature placental separation.
The first stage of labour can be managed as for singleton pregnancy and vaginal birth of twin 1 usually proceeds as normal. Immediately after the birth of the first twin, it is important to
• continuously monitor the fetal heart rate of twin 2 to ensure fetal wellbeing. Where there is concern, a vacuum extraction or breech extraction can be performed without resorting to Caesarean section
• determine the position of twin two. If longitudinal, normal delivery will usually result after rupture of the second amniotic sac. If transverse, internal podalic version or external version may be used to establish a longitudinal lie
• contractions usually abate after the birth of the first twin. An intravenous oxytocin infusion is usually required to help deliver the second twin. The second twin should deliver within 30 minutes of the first twin.
The third stage of labour should be actively managed. Higher order multiple pregnancy
The outcomes of higher order multiples (three or more) are significantly worse than twin or singleton pregnancies.
Although previously a relatively common occurrence with in vitro fertilization (IVF) treatment, the Human Fertilization and Embryology Authority has limited the maximum number of embryos (two) transferred per cycle of IVF in order to reduce the number of multiple pregnancies.
Multifetal pregnancy reduction may be carried out, but carries a risk of miscarriage (5–8%) as well as considerable ethical problems. It is usually performed after the nuchal translucency scan at 12 weeks with ultrasound-guided injection of potassium chloride into the selected fetuses to leave a twin pregnancy. Further reading
Taylor MJ. The management of multiple pregnancy. Early Hum Dev 2006;82:365–70. Patient resources
www.tamba.org.uk
Oligohydramnios Definition
Oligohydramnios is defined as reduced amniotic fluid volume (AFV) for a given gestational age. Oligohydramnios is secondary to either an excess loss of fluid, or a decrease in fetal urine production or excretion. Epidemiology
Oligohydramnios is a complication in ~5% of all pregnancies, and severe oligohydramnios in ~1% of pregnancies. Oligohydramnios is more common in pregnancies beyond term, because the AFV normally decreases at term. It complicates as many as 12% of pregnancies that last beyond 41 weeks. Differential diagnosis
Physiological changes in amniotic fluid volume
Amniotic fluid volume decreases with advancing gestation, especially in the third trimester. The significance of reduced amniotic fluid volume in late pregnancy is debatable.
Estimation of amniotic fluid volume
Measurement of the amniotic fluid index (AFI) or single deepest pool is only a proxy for the amniotic fluid volume. This technique has a poor specificity and sensitivity for the diagnosis of oligohydramnios. Pathology/aetiology
Uteroplacental insufficiency
Oligohydramnios is an early feature of uteroplacental insufficiency and is associated with decreased fetal biometry particularly the abdominal circumference. Other ultrasound and Doppler features of fetal growth restriction aid in the confirmation of uteroplacental insufficiency as the cause for the reduced amniotic fluid volume.
Amniotic membrane rupture
The maternal history of persistent vaginal loss and dampness would suggest a diagnosis of prelabour membrane rupture. This is often associated with anhydramnios rather than oligohydramnios. The finding of normal amniotic fluid volume or oligohydramnios, however, does not exclude this diagnosis.
Abnormal fetal renal function
Bilateral renal agenesis, polycystic kidney disease, multicystic dysplasia and obstructive uropathy (bladder outflow obstruction) characteristically present with anhydramnios. However, early on in the process of renal impairment, transient oligohydramnios may be evident.
Post-term gestation
Although oligohydramnios may be physiological, it may be related to placental failure that is known to occur more frequently at this gestation. Oligohydramnios in the post-term patient is associated with more fetal decelerations, a higher incidence of meconium-stained fluid, and an increased risk for caesarean delivery. Anhydramnios
Anhydramnios is defined as absent amniotic fluid at any gestation. The finding of anhydramnios in the first and second trimesters is usually associated with a poor prognosis because of the subsequent development of lethal fetal pulmonary hypoplasia. The presence of anhydramnios is also associated with joint contractures such as talipes (club foot). Clinical approach Assessment of amniotic fluid volume
The amniotic fluid volume increases from approximately 250 mL at 16 weeks to 1000 mL at 34 weeks, declining thereafter to approximately 800 mL at term. The amniotic fluid volume reflects both the maternal and fetal status and is altered in many physiological and pathological conditions. There are three methods for assessing amniotic fluid volume.
Subjective assessment
It is possible to classify amniotic fluid volume into broad categories such as absent, low, normal, increased, and excessive. This method has proved impossible to standardize in clinical and research terms.
Single deepest pool
The size of the deepest, cord-free pool of amniotic fluid is assessed using ultrasound. A 2–3 cm pool is considered acceptable in normal pregnancy.
Amniotic fluid index
Using the maternal umbilicus as a reference point, the abdomen is divided into four quarters. Using ultrasound, the largest vertical pool depth is recorded in each quadrant. The sum of these measurements represents the amniotic fluid index (AFI). Although the AFI is known to vary with gestational age, an AFI of amniotic fluid volume is of unquestionable value. However, a practical and reproducible technique for the accurate assessment of amniotic fluid volume is yet to be introduced into clinical practice. Management
Anhydramnios as a consequence of renal pathology or early/midtrimester membrane rupture is frequently associated with a poor prognosis. If oligohydramnios is due to uteroplacental insufficiency, the management will depend on the severity of growth restriction and the gestation of the pregnancy.
Oligohydramnios of unknown aetiology on the other hand is of dubious clinical significance. Given the poor reproducibility of subjective and objective amniotic fluid estimations, one could question the value of reporting this when present as an isolated finding. The exception to this recommendation would appear to be prolonged or post-term pregnancy, where reduced amniotic fluid volume may be associated with poorer fetal and neonatal outcomes.
Amnio-infusion
Increasing the amount of fluid within the amniotic cavity can be accomplished during delivery with the use of amnioinfusion (instillation of warm sodium chloride solution into the cervix). This procedure increases the amount of fluid around the umbilical cord, which has been shown to decrease the frequency and severity of fetal heart decelerations in labour. Unfortunately, this is not reflected in lower caesarean section rates or improved neonatal outcomes.
Vesico-amniotic shunts
Vesico-amniotic shunts may be used to divert fetal urine to the amniotic fluid cavity in patients with a fetal obstructive uropathy. Although it is effective in reversing oligohydramnios, its ability to achieve sustainable neonatal renal and pulmonary function remains to be established. Further reading
Sherer DM, Langer O. Oligohydramnios: use and misuse in clinical management. Ultrasound Obstet Gynecol 2001;18:411–19.
Placental abnormalities
The placenta is an organ with both maternal and fetal origins. It is composed of functional units called villi, through which the mother and fetus exchange nutrients and waste products. The placenta is also implicated in the mechanism of immune avoidance by the fetus and undertakes an important endocrine role in pregnancy. Normal variation in placental morphology
Succenturiate lobe
This refers to the presence of one or more accessory lobes of the placenta. Making the diagnosis is important as it is possible to have a fundal placenta together with a lowlying succenturiate lobe where the internal cervical os is covered by placenta (placenta praevia) or membrane with blood vessels within them (vasa praevia). These can be life-threatening conditions to the mother and baby. Succenturiate lobes may also be retained after delivery and become a cause of postpartum haemorrhage or infection.
Placental lakes
These are areas within the bulk of the placenta that are filled with slowly moving blood. There is a relationship between the presence of placental lakes and fetal growth restriction, but it is weak enough to be of little clinical significance.
Placental cysts
These are found just below the chorionic plate. They are though to represent deposition of fibrin in the intervillous space and are of no apparent significance.
Placental maturation
This is a classification of the normal changes that occur in the placenta during the course of a pregnancy; it is often known as Grannum grading. Originally it was suggested that increasing Grannum grades were associated with placental dysfunction. However, grading is no longer routinely used because of other, more accurate ways of assessing fetal wellbeing and placental function. Placental abnormalities
Chorioangioma
This is a benign vascular tumour of the placenta (Fig. 7.22.1). Such tumours vary both in appearance and size. If the tumours have numerous feeding vessels, are large or are close to the umbilical cord insertion, they may result in significant fetal arteriovenous shunting. This can cause fetal hyperdynamic circulation, high-output fetal cardiac failure and the development of polyhydramnios and hydrops fetalis. Occasionally they can be associated with fetal growth restriction, presumably because they reduce the effective functional capacity of the placenta.
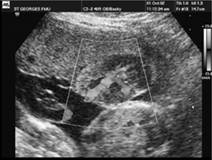
Fig. 7.22.1 Ultrasound image showing a placental chorioangioma, with colour Doppler blood flow within it. See also colour plate section.
Circumvillate placenta
In approximately 1% of cases, there is a small central chorionic area inside a paler thick ring of membranes on the fetal side of the placenta. This is associated with an increased rate of antepartum bleeding, prematurity, abruption, multiparity, and perinatal death.
Other placental problems
For information on placenta praevia, accreta, abruption, and retained placenta, see Chapter 10, Care in labour. Umbilical cord abnormalities
Two-vessel cord
The absence of one umbilical artery is relatively common, with a reported incidence of approximately 1%. Associated malformations are thought to be present in as many as 50% of cases, with cardiac and renal being the systems most commonly affected. The risk of an underlying fetal chromosomal abnormality is unaltered in a previously screened population. The likelihood of the pregnancy being affected by intrauterine growth restriction is also thought to be increased making follow-up growth scans part of the routine management.
Marginal insertion of cord (Battledore)
Where the cord has a marginal rather than central insertion to the placenta. This has no clinical significance.
Velamentous cord insertion
The placental vessels run and divide in the membrane before reaching the cord. This is only of clinical significance if the vessels cross the lower pole of the uterus (vasa praevia, see Chapter 10, Care in Labour). With the latter finding, there is high risk of fetal haemorrhage and death at rupture of membranes. It can be diagnosed prenatally by ultrasound examination and managed by elective Caesarean section.
Coiling of the umbilical cord
Reduced coiling of the umbilical cord is associated with uteroplacental insufficiency. Hypercoiling of the cord is also associated with poorer neonatal outcomes, because of associated fetal abnormalities. There is no established, reproducible method of assessing umbilical vessel coiling reliably. Hence coiling is not used clinically to indicate the need for regular scans or detailed fetal assessment.
Umbilical cord masses
These may be primary in nature, originating from the remnants of the allantoic or vitelline duct. The use of colour Doppler will help differentiate cord masses of vascular origin and their relationship to the umbilical vessels.
Abnormal length of cord
A long cord (> 100 cm) is associated with increased risk of fetal entanglement, knots and prolapse of the cord. A short cord (< 40 cm) may be associated with a poorly active fetus, Down’s syndrome, cord rupture, breech position, prolonged second stage, uterine inversion and abruption. Further reading
Benirschke K, Kaufmann P. Pathology of the human placenta, 2nd edn. New York: Springer-Verlag 1990.
Kaplan CG. Postpartum examination of the placenta. Clin Obstet Gynecol 1996;39:535–48.
Polyhydramnios Definition
Subjectively assessed, or amniotic fluid index above the 95th centile for gestational age, or a maximum vertical pool length of ≥8 cm or AFI ≤24 (Fig. 7.23.1). Epidemiology
It complicates 0.5–1% of pregnancies. Aetiology
Excessive amniotic fluid is the result of either excessive liquor production or reduced clearance. Mild idiopathic polydramnios with no identifiable cause accounts for 55% of cases.
Diabetes in pregnancy is the commonest identifiable cause (25%).
Early onset polyhydramnios with associated structural anomalies (18%) should prompt consideration of chromosomal syndromes.
Increased liquor production
• Maternal diabetes
• Twin pregnancy (mostly monochorionic with twin–twin transfusion syndrome)
• Placental tumour i.e. chorioangioma (rare)
• Fetal macrosomia.
Decreased swallowing
• GastrointestinaI atresia
• tracheoesophageal fistula
• oesophageal atresia
• duodenal atresia
• Chest malformations
• diaphragmatic hernia
• Neuromuscular disorder
• myotonic dystrophy
• anencephaly
• Lethal skeletal dysplasia.
Hydrops
• Fetal anaemia (parvovirus infection, red cell alloimmunization)
• Cardiac arrhythmia
• Other causes (see Chapter 7.17). Prognosis
In the presence of structural anomalies or chromosomal syndromes, prognosis depends on the underlying pathology.

Fig. 7.23.1 Amniotic fluid index in normal pregnancy from 16 to 42 weeks’ gestation (lines represent the 5th, 50th, and 95th centiles) modified from Moore TR, Cayle JE.
Idiopathic polyhydramnios is not associated with an increased rate of preterm delivery. However, perinatal mortality appears increased two- to fivefold compared with healthy control pregnancies. Clinical approach
Diagnosis History
• Presenting complaint
• abdominal discomfort
• dyspnoea
• Current pregnancy
• gestational diabetes
• dipstick glycosuria
• maternal red cell antibodies
• Risk factors
• high BMI
• previous pregnancy affected
• pre-existing diabetes
• previous macrosomia
• previous structural malformation or genetic syndrome.
Ultrasound findings
• A detailed structural survey should be performed to rule out congenital malformations:
• markers for chromosomal defects
• absent stomach bubble
• ‘double bubble’ sign
• diaphragmatic hernia.
• Abnormal posture or absent movements may suggest neuromuscular disorder.
• Abnormal size or shape of long bones may suggest lethal skeletal dysplasia.
• Middle cerebral artery (MCA) Doppler assessing peak systolic velocity to diagnose fetal anaemia.
• Fetal cardiac scan if there is evidence of cardiac arrhythmia or hydrops.
• Placental survey for vascular masses.
Investigations
• Glucose tolerance testing
• Red cell antibodies (if not previously done)
• Parvovirus serology, if raised MCA peak systolic velocity on Doppler
• Maternal screening for toxoplasamosis and cytomegalovirus
• In the presence of structural anomalies or hydrops consider amniocentesis for karyotyping
• In the presence of hydrops for full list of investigations see Chapter 7.17. Counselling and management
In the presence of associated structural anomalies, counselling will be guided by the underlying condition. In mild idiopathic cases, guarded reassurance may be given. However, it should be explained that it is not possible to exclude oesophageal atresia or tracheo-oesophageal fistula if one is not seen on ultrasound: postnatal assessment of the neonate is always indicated.
Aetiological treatment
• In diabetic pregnancies presence of worsening polyhydramnios is a sign of poor glycaemic control. Frequent glucose monitoring and increased insulin requirement is indicated.
• In TTS the options of laser ablation of arteriovenous anastomoses, amnio drainage, or delivery may be considered depending on the gestational age, stage, and severity of disease.
• Fetal anaemia may be managed with cordocentesis and intrauterine fetal transfusion.
Symptomatic treatment
• Serial amnio reductions may become necessary, especially in upper gut obstructions, in order to treat maternal symptoms or prevent preterm delivery.
• Indomethacin may reduce fetal urine production through direct renal effect and may also exert a tocolytic effect. However, it is associated with premature closure of the ductus arteriosus and is therefore not recommended.
• Steroid cover should be considered in cases of severe tense polyhydramnios at less than 34 weeks gestation when the risk of preterm delivery is significant.
• In women with severe discomfort and respiratory compromise induction of labour at or beyond 37 weeks may be indicated. Further reading
Desmedt EJ, Henry OA, Beischer NA. Polyhydramnios and associated maternal and fetal complications in singleton pregnancies. Br J Obstet Gynaecol 1990;97:1115–22.
Magann EF, Chauhan SP, Doherty DA, et al. A review of idiopathic hydramnios and pregnancy outcomes. Obstet Gynecol Surv 2007;62:795–802.
Williams K. Amniotic fluid assessment. Obstet Gynecol Surv
1993;48:795–800. Further reading
Thilaganathan B, Sairam S, Papageorghiou AT, Bhide A. Problem based obstetric ultrasound, 1st edn. Informa Healthcare 2007. Internet resources
www.emedicine.com (needs free registration) Patient resources
www.babycentre.co.uk/pregnancy/complications/polyhydramnios/
Red blood cell isoimmunization Definition
Fetomaternal blood group incompatibility may result in the development of maternal antibodies, which cross the placenta, become attached to the fetal red cells, and cause their destruction. Epidemiology
There are at least 100 red blood cell surface antigens and the development of maternal antibodies to approximately 30 of these can lead to fetal haemolytic disease. However, the vast majority of cases are the result of either ABO or rhesus incompatibility.
ABO fetomaternal incompatibility occurs in about 20% of all pregnancies, but fetal haemolysis occurs in less than 2% of cases and this is usually mild. The antibodies are usually immunoglobulin (Ig)M, which do not cross the placenta and no special antenatal measures are necessary for the management of these pregnancies.
The commonest antibody causing severe fetal haemolysis is anti-D. Other antibodies causing fetal haemolysis are anti-C, anti-Kell and anti-E and rarely antibodies such as anti-M, anti-N, anti-S. Pregnancies complicated by the presence of these antibodies should be managed in the same way as those of Rhesus (D) incompatibility, including maternal antibody level estimations, Doppler studies, cordocentesis, fetal blood transfusion, and early delivery.
A Rhesus (D)-positive infant is born to a Rhesus (D)-negative mother in 10% of Caucasian and 3% of Black pregnancies. Clinical management
Rh-negative women with no Rh antibodies
Anti-D administration is effective in preventing immunization in a Rhesus-negative woman but has no effect if the woman is already isoimmunized. The administration of 100 μ g of anti-D IgG to a mother, will neutralize at least 4 mL of Rhesus (D)-positive fetal erythrocytes that might have entered her circulation. This is equivalent to a Kleihauer count of 80 fetal cells per 50 low-power fields. Routine antenatal anti-D prophylaxis with anti-D IgG is recommended for all pregnant women who are Rh D negative and who are not known to be sensitized, and within 72 hours of the delivery of a Rhesus-positive baby and also after termination, miscarriage, antepartum haemorrhage, amniocentesis, or other potentially immunizing episodes. Routine prophylaxis is commonly given as two doses of 500–1650 IU (one at 28 weeks and one at 34 weeks’ gestation), or as a single dose of 1500 IU either at 28 or 30 weeks of gestation.
At the first antenatal visit the ABO and rhesus blood group should be determined in all patients. Furthermore, screening for other atypical antibodies should be performed. If the woman is Rhesus negative, the Rhesus status of the partner is determined. If he is also Rhesus negative then the fetus must be Rhesus negative and the mother is not at risk of Rhesus isoimmunization.
If the father is Rhesus positive then further maternal antibody screening should be undertaken at 20 weeks and at 4-week intervals thereafter until delivery.
After delivery, the blood group and rhesus status of the fetus and the direct Coombs test for the presence of antibodies on the fetal erythrocytes should be performed. If the mother has no antibodies and the baby is Rhesus D positive, anti-D IgG should be given.
Red cell isoimmunized pregnancies
In the management of red cell isoimmunized pregnancies the aims are
• to predict whether the fetus is severely affected
• to correct the fetal anaemia by intrauterine blood transfusion
• to deliver the baby at the optimal time by balancing the risks of prematurity versus intrauterine transfusion.
The only accurate method of assessing the severity of fetal anaemia is by fetal blood sampling. However, cordocentesis should only be undertaken if there is a strong suspicion that the fetus is severely affected because the procedure itself can cause miscarriage and it can also cause fetomaternal haemorrhage thereby exacerbating the severity of the disease.
The fetal haemoglobin concentration normally increases with gestation from a mean of 11 g/dL at 18 weeks to 14 g/dL at 38 weeks and 1 SD is about 1 g/dL. Mild anaemia is defined by a haemoglobin deficit of 2–4 g/dL, moderate disease by a deficit of 4–6 g/dL, and severe disease by a deficit of more than 6 g/dL. When the fetal haemoglobin concentration deficit exceeds 6 g/dL hydrops fetalis develops.
In cases where the father is homozygous for the Rhesus (D) antigen, the fetus is Rhesus positive. If the father is heterozygous the fetus could be negative, and in these cases the fetal Rhesus status can now be determined accurately by examination of the cell-free fetal DNA in maternal plasma.
Assessment of the severity of fetal haemolysis should be based on:
• the history of previous affected pregnancies
• the levels of maternal haemolytic antibodies
• ultrasonographic examination for the detection of ascites
• Doppler studies for diagnosis of a hyperdynamic circulation. Cordocentesis can safely be reserved only for those pregnancies demonstrating increased fetal middle cerebral artery peak systolic velocity (MCA-PSV); in more than 95% of the severely anaemic fetuses the MCA-PSV is more than 1.5 multiples of the median above the normal median for gestation. This high sensitivity can be achieved with a relatively low false-positive rate and therefore avoidance or delay in invasive testing in more than 80% of isoimmunized pregnancies with high maternal serum antibody concentration.
For patients with a previous red blood cell isoimmunization-affected pregnancy, it should be aimed to perform the first ultrasound scan and Doppler studies at approximately 10 weeks before the time of the earliest previous fetal or neonatal death, fetal transfusion, or birth of a severely affected baby, but not before 17–18 weeks. Fetal death or the development of hydrops do not occur before this gestation, presumably because the fetal reticuloendothelial system is too immature to result in destruction of antibody coated erythrocytes. Assessment should be carried out at intervals of 1–2 weeks and cordocentesis need only be performed if there is fetal ascites or if the fetal MCA-PSV is more than 1.5 MoM above the normal mean for gestation.
In patients that had no or mildly affected previous pregnancies the maternal haemolytic antibody levels should be measured at 2–3 weekly intervals from 17 weeks onwards. When the antibody concentrations are persistently below 15 IU/mL (equivalent to a dilution of 1 in 128), the degree of fetal haemolysis is insignificant or mild and delivery can be delayed until term. If the antibody levels are higher than 15 IU/mL the disease may be severe and the fetus should be assessed by ultrasound and Doppler examinations at 1-week intervals and cordocentesis should be considered in those that develop ascites or a high MCA-PSV.
Fetal blood transfusions
In very severe disease requiring fetal transfusions before 20 weeks the intraperitoneal approach is safer, but after 20 weeks the intravascular approach is preferred.
Intraperitoneal transfusions are performed by the ultrasound guided insertion of a 20-gauge needle into the amniotic cavity. The transducer is aligned allowing a view transverse to the fetal abdomen and perpendicular to the course of the needle, which is directed towards the anterolateral aspect of the fetal abdomen between the bladder and umbilicus and then thrust forward into the peritoneal cavity. The hub of the needle is connected to a syringe through a three-way tap. Donor blood is drawn into the syringe and infused manually at 10 mL/minute, during which time the fetal heart rate is monitored continuously by ultrasound. The total volume of blood transfused is 5 mL for 16–18 weeks, 10 mL for 19–21 weeks, and 10 mL per week of gestation after 21 weeks. This avoids over transfusion, which would cause the intraperitoneal pressure to rise above the umbilical venous pressure, obstruct the placental circulation, and result in fetal death.
Intravascular transfusions are given by cordocentesis using a 20-gauge needle. Where the placenta is anterior the needle is advanced through the placenta and into the base of the umbilical cord. When the placenta is posterior, the needle is introduced into the amniotic cavity and the cord punctured near the placental insertion. The stylet of the needle is then removed, a heparinized syringe attached to the needle hub, and pure fetal blood aspirated. The fetal haemoglobin concentration is determined and if this is below the normal range, the tip of the needle is kept in the lumen of the umbilical cord vessel and fresh, packed, Rhesus-negative blood compatible with that of the mother is infused manually into the fetal circulation. The sono-graphically detectable echogenicity in the umbilical cord produced by the infused blood allows identification of the punctured vessel. The fetal heart rate and the flow of the infused blood are monitored continually throughout the procedure by ultrasonography. At the end of the transfusion a blood sample is aspirated for determination of the final haemoglobin concentration. The volume of donor blood is calculated by considering the pretransfusion fetal haemoglobin concentration, the haemoglobin of the transfused blood, the desired post-transfusion haemoglobin, and the normal mean fetoplacental blood volume for that gestation.
Subsequent transfusions are given at 1–3-week intervals and the babies are delivered at 35–40 weeks. Prognosis
The survival rate of red cell-isoimmunized pregnancies treated with cordocentesis is more than 90%. Further reading
Hecher K, Snijders R, Campbell S, Nicolaides KH. Fetal venous, intracardiac, and arterial blood flow measurements in intrauterine growth retardation: relationship with fetal blood gases. Am J Obstet Gynecol 1995;173:10–5.
Mari G. Noninvasive diagnosis by Doppler ultrasonography of fetal anemia due to maternal red-cell alloimmunization. N Eng J Med 2000;342:9–14.
Nicolaides KH, Rodeck CH. Maternal serum anti-D antibody concentration and assessment of rhesus isoimmunisation. BMJ 1992;304:1155–6.
Nicolaides KH, Soothill PW, Clewell WH, et al. Fetal haemoglobin measurement in the assessment of red cell isoimmunisation. Lancet 1988;1:1073–5.
Nicolaides KH, Soothill PW, Rodeck CH, Clewell W. Rh disease: intravascular fetal blood transfusion by cordocentesis. Fetal Therapy 1986;1:185–92.
Rightmire DA, Nicolaides KH, Rodeck CH, Campbell S. Fetal blood velocities in Rh isoimmunization: relationship to gestational age and to fetal haematocrit. Obstet Gynecol 1986;68:233–6.
Vyas S, Nicolaides KH, Campbell S. Doppler examination of the middle cerebral artery in anemic fetuses. Am J Obstet Gynecol 1990;162:1066–8 Internet resources
www.nice.org.uk/TA156 Patient resources
www.patient.co.uk/doctor/Haemolytic-Disease-of-the-Newborn.htm
Screening for fetal aneuploidy
There is a fundamental difference between antenatal screening and prenatal diagnosis for congenital abnormalities.
• Screening is limited to the identification of those at high enough risk to justify further investigation.
• Diagnosis is definitive but involves invasive procedures, mainly chorionic villus sampling (CVS) and amniocentesis, which carry a risk of miscarriage.
Currently, screening assesses risk of fetal aneuploidy by combining information on maternal age and multiple markers in maternal serum and ultrasound. Markers are continuous variables with a distribution of values that is, on average, higher or lower in affected pregnancies. Aneuploidy
This is a common event in pregnancy but most affected embryos miscarry spontaneously early in the first trimester, many of them even before there are clinical signs of pregnancy. Those that survive into the second trimester also experience high late-intrauterine mortality and increased risk of infant death.
Birth prevalence in the absence of prenatal diagnosis and therapeutic abortion:
• Down’s syndrome (DS): 1–2 per 1000, depending on the maternal age distribution, of which 95% are non-disjunction, 4% translocation, 1% mosaic
• Edwards’ syndrome: 1/9 less frequent
• Patau’s syndrome: 1/20 less frequent
• Turner’s syndrome and other sex chromosome aneuploidies are more common but relatively benign.
DS screening
This chapter is mainly concerned with DS risk assessment. Increasingly Edwards’ syndrome risks are also included on DS screening reports (see below).
Natural history
The life expectancy is considerably greater than in the past, although precise estimates are difficult to derive. However, it remains the most common known cause of severe mental handicap. In addition, many adults may experience cognitive deficits due to pathological changes in the brain normally associated with Alzheimer’s disease.
Prescreening risk
This can be expressed as a rate 1 in 1/p, where p is prevalence; or as an odds p:(1–p) or 1:(1–p)/p.
• Trimester: Most centres quote the risk at term, but some give the mid- or first trimester risks, which are respectively about one-quarter higher and double.
• Maternal age: This is the most important prescreening factor. Risk changes little from 1 in 1500 at age 20 to 1 in 900 at 30 and then increases rapidly to 1 in 30 at 45.
• Paternal age: Contributes little or no additional risk.
• Family history: Unless there is a familial translocation or mosaicism, only a previous affected pregnancy is relevant. This increases risk to 1 in 200 at 30 and 1 in 25 at 45. Markers
Of the more than 50 maternal blood, maternal urine or ultrasound markers, currently seven are widely used: maternal serum human chorionic gonadotrophin (hCG), the free β-subunit of hCG, α-fetoprotein (AFP), unconjugated oestriol (uE3), inhibin A and pregnancy-associated plasma protein (PAPP)-A, and ultrasound measurement of fetal nuchal translucency (NT), the single most discriminatory marker, albeit limited to 11–13 weeks of gestation and optimal at 11 weeks.
Adjustments
• Gestation: all the widely used markers change with gestation. Therefore, results are expressed as multiples of the normal gestation-specific median (MoM) using a local regression equation.
• Operator: NT can be difficult to quality control and some centres use operator-specific regression equations (Logghe et al. 1995).
• Maternal weight: all serum marker levels decline with weight and MoMs are adjusted for this.
• Ethnicity: women of Afro-Caribbean origin have PAPP-A levels about 50% higher. Either ethnic-specific normal medians or a multiplication factor is needed. Other markers are altered less as are those in Oriental and Asian origin women.
• Smoking: PAPP-A levels are reduced in smokers; hCG and free β-hCG levels are also reduced but only in the second trimester. Information collected during pregnancy on daily intake is often inaccurate. Hence, although some centres adjust for smoking per se, adjustment for intake is not recommended.
• Assisted reproduction: PAPP-A, hCG and free β-hCG levels are raised in pregnancies conceived by IVF, ICSI, intrauterine insemination or following ovulation induction alone. However, there is considerable between-study heterogeneity, possibly due to the method of gestational assessment, the cause of infertility, and treatment. Hence, adjustment is not generally applied.
Frequency distributions
After log transformation each of the widely used markers, both in DS and unaffected pregnancies, follows a Gaussian distribution over most of the range. Moreover, all combinations of two or more markers follow a multivariate Gaussian distribution.
The distribution parameters are the means and standard deviations of the individual markers and the correlation coefficients between markers. They are probably best derived by meta-analysis. Risk calculation and interpretation
The prescreening risk, expressed as odds is multiplied by a factor known as the ‘likelihood ratio’ (LR) derived from the marker profile. The resulting odds are then re-expressed as a rate. This posterior risk is then compared with a fixed cut-off risk. If the risk is greater than the cut-off the result is regarded as positive, otherwise it is negative.
The likelihood ratio for a single marker is calculated by the ratio of the heights of the two overlapping distributions at the specific level. For extreme results that fall beyond the point where the data fit a Gaussian distribution, it is standard practice to use the LR at the end of the acceptable range. For more than one marker the heights of multivariate log Gaussian distributions are used.
Predicting screening performance
This is determined by the detection rate (DR), the proportion of DS pregnancies referred for prenatal diagnosis and the false-positive rate (FPR).
The most robust predictions are from statistical modelling. The same Gaussian model used to calculate LR is used to generate distributions of risk in DS and unaffected pregnancies. The maternal age distribution can either be derived nationally or modelled (e.g. Gaussian, mean 27 and SD 5.5 years).
Either the DR is predicted for a fixed FPR (e.g. 1% or 5%), the FPR for a fixed DR (e.g. 75% or 85%), or DR and FPR for a given cut-off (e.g. 1 in 250 at term or 1 in 270 at midtrimester). Principal strategies
Initially DS screening was carried out at 15–19 weeks to utilize the existing neural tube defect screening programme. Over the last decade there has been a gradual realization of the benefits of moving from the second to the first trimester. Today strategies are emerging that use both first and second trimester markers sequentially.
Second trimester (1T) tests
• Double: AFP and hCG or free β-hCG
• Triple: double plus uE3
• Quadruple: triple plus inhibin A.
First trimester (2T) tests
• NT alone
• Combined: NT, PAPP-A and free β-hCG.
Sequential tests
• Serum-integrated: first trimester PAPP-A plus second trimester quadruple markers. The first trimester result is not disclosed until the test is complete.
• Integrated: serum integrated plus NT.
• Stepwise: first trimester combined markers but using a very high cut-off; women with risks below the cut-off are offered second trimester quadruple markers and risk revision using all markers.
• Contingent: stepwise except that only women with borderline risks are offered the second stage.
Some regard non-disclosure of the two integrated tests to be unethical, or at least impractical due to the difficulty for the professional not to act on intermediate findings which would of themselves be abnormal, particularly the NT, and any increase in detection is paid for by sacrificing early diagnosis and reassurance.
Another approach, the ‘independent sequential’ test, albeit statistically invalid, is being practised to some degree by default, namely to carry out a combined test followed by a quadruple test and to calculate separate risks from each.
Model predicted DR for 5% FPR
Using parameters from one meta-analysis:
• double: 56% with hCG and 61% with free β-hCG
• triple: 60% and 65%
• quadruple: 67% and 71%
• NT alone: 71% at 13 weeks and 77% at 11 weeks
• combined: 81/80% and 84/87% with first trimester hCG/free- B hCG
• serum integrated: 73% and 78%
• integrated: 89% and 93%
• stepwise: 91/91% and 94/94% with first trimester hCG/free β-hCG
• contingent: 88/88% and 90/92% with first trimester hCG/free β-hCG
• independent: 84% for all weeks and hCG types.
These rates are useful for health policy planning. However, for individual choice the maternal age-specific DR and FPR is needed; for a given risk cut-off, both increase with age.
Prospective intervention studies
In such studies the observed DR overestimates the actual value. This arises because some DS pregnancies terminated following a high-risk result are destined to miscarry anyway, whereas non-viable DS pregnancies with low-risk results are lost to follow-up. The DR can be corrected using trimester-specific viability factors.
Meta-analysis of published studies confirms predictions for the well established strategies:
• double: 234 000 tests, corrected DR 59%, FPR 5.0%
• triple: 612 000, 67%, 6.5%
• quadruple: 86 000, 79%, 7.4%
• NT alone: 104 000, 72%, 8.4%
• combined: 145 000, 81%, 5.9%.
So far there are insufficient published sequential test studies, but head-to-head retrospective comparison within the intervention FaSTER trial confirmed that integrated, step-wise and contingent tests had similar DR. Possible future strategies
Contingent combined test
Where there is limited availability of quality NT, the combined test serum markers are tested on all women and NT is limited to those with high post-test DS risks. Predicted DR is 82% when one-third have NT.
Three-stage contingent test
The small loss of detection with the contingent combined test can be completely recouped by the contingent determination of second trimester serum markers just like a standard sequential contingent test.
Additional first trimester serum markers
AFP, uE3 and inhibin are weak first trimester markers but can be added to the combined test. Another marker, A disintegrin and metalloprotease (ADAM)12s, might be discriminatory prior to 9 weeks gestation.
Additional first trimester ultrasound markers
Four can be determined at the same time as NT: absence of the fetal nasal bone (NB), abnormal blood flow in the ductus venosus, tricuspid regurgitation, and the frontalmaxillary facial angle, requiring three-dimensional scanning. Few centres are sufficiently proficient to determine these routinely although NB is now becoming more available.
Based on meta-analysis the LRs for absent and present NB are 49 and 0.31, although different values are needed according to CRL, NT, and ethnicity.
Modelling predicts a substantial increase in DR:
• NT and NB: 87% and 90% at 13 and 11 weeks.
• combined with NB: 92/91% and 93/94% with hCG/free β-hCG.
• contingent with NB: 91/90% and 92/93%.
First trimester contingent test
Where there is limited availability of additional first trimester markers, refer those with borderline contingent test risks for specialist ultrasound and risk re-evaluation.
Soft markers
The results of an 18–22-week ‘anomaly scan’ are sometimes used in the post hoc modification of risk among women considering amniocentesis. Major anomalies are risk factors as are so-called ‘soft’ markers: increased nuchal skinfold (NF), short femur and humerus lengths, hydronephrosis, echogenic intracardiac focus, and echogenic bowel.
These markers could also be formally incorporated into routine screening policies outside specialist centres. NF is most suited to this together with facial profile markers: nasal bone length (NBL) and prenasal thickness (PT). All three are all continuous variables with good Gaussian fit.
Second trimester combined test
Uses quadruple test markers together with ultrasound NF, NBL and PT. Modelling predicts the following DRs (Cuckle and Benn 2009):
• quad and NF: 78/80% with hCG/free β-hCG
• quad, NF, and NBL: 83/84%
• quad, NF, NBL, and PT: 92/93%.
Repeat measures
Markers which are more discriminatory at one gestation than another (e.g. PAPP-A) could be measured sequentially to capture a changing profile. Modelling predicts a large benefit. Twins
Serum marker levels are approximately double those of singletons except for uE3, where they are two-thirds higher. In monozygotic twins, determined by ultrasound examination of the intertwine membrane, DR is similar to singletons, but in dizygotic twins the normal co-twin ‘masks’ the DS serum marker profile and DR is reduced.
If there is a ‘vanishing twin the first trimester serum marker profile may be altered and some advocate delayed testing. Other disorders
• Edwards’ syndrome: four-fifths are detected through high DS risk in a combined test and one-third in second trimester tests. Explicit use of Edwards’ syndrome risk changes the DR for the combined test little but increase to one-half for second trimester tests.
• Neural tube defects: about 90% of cases of anencephaly and 75% of spina bifida have raised AFP after 15 weeks. However, there is a much higher DR with the anomaly scan where, in the BPD view, the ‘lemon and banana’ signs are seen.
• X-linked ichthyosis: identified with low uE3.
• Smith–Lemli–Opitz syndrome: low uE3 and other 2T markers.
• Cornelia de Lange syndrome: low PAPP-A and possibly free β-hCG and inhibin.
• Cardiac abnormalities: increased NT with normal karyotype is an indication for detailed fetal heart examination.
• Molar pregnancy or placental dysplasia: undetectable AFP and uE3 or very low PAPP-A.
• Fetal demise: low PAPP-A and low or high free β-hCG.
• Pre-eclampsia: reduced PAPP-A and increased inhibin. References
Arbuzova S, Cuckle H, Mueller R, Sehmi I. Familial Down syndrome, evidence supporting cytoplasmic inheritance. Clin Genet 2001;60:456–62.
Cicero S, Rembouskos G, Vandecruys H, et al. Likelihood ratio for trisomy 21 in fetuses with absent nasal bone at the11–14-week scan. Ultrasound Obstet Gynecol 2004;23:218–23.
Cuckle H. Down’s syndrome screening in twins. J Me Screening 1998;5:3–4.
Cuckle H, Benn P. Multianalyte maternal serum screening for chromosomal defects. In: Genetic disorders and the fetus: diagnosis, prevention and treatment, 6th edn, Milunsky A. (ed.). Baltimore: Johns Hopkins University Press 2009.
Cuckle HS, Wald NJ, Thompson SG. Estimating a woman’s risk of having a pregnancy associated with Down’s syndrome using her age and serum alpha-fetoprotein level. Br J Obstet Gynaecol 1987;94:387–402.
Cuckle H, Aitken D, Goodburn S, et al. UK National Down’s Syndrome Screening Programme, Laboratory Advisory Group. Age-standardisation when target setting and auditing performance of Down syndrome screening programmes. Prenat Diagn 2004;24:851–6.
Logghe H, Cuckle H, Sehmi I. Centre-specific ultrasound nuchal translucency medians needed for Down’s syndrome screening. Prenat Diagn 1995;235:389–92.
Royston P, Thompson SG. Model-based screening by risk with application to Down’s syndrome. Stats Med 1992;11:257–68.
Wright D, Bradbury I. Repeated measures screening for Down’s syndrome. Br J Obstet Gynaecol 2005;112:80–3.
Symphyseal fundal height Definition
The symphyseal fundal height (SFH) measurement is a measurement of the longitudinal distance of the pregnant abdomen from the symphysis pubis to the uterine fundus. It is used as a screening tool for abnormal fetal growth in late second and third trimester antenatal care, primarily to detect intrauterine growth restriction, but also as to identify macrosomia, multifetal pregnancy, and polyhydramnios. Background
One of the main targets of antenatal care is the timely detection of abnormal fetal growth, as growth abnormalities may lead to adverse pregnancy outcomes, including perinatal death. The intrauterine growth restriction (IUGR) fetus is at increased risk in prolonged pregnancy and in labour, and may warrant intervention. The macrosomic fetus and the pregnant woman are both at increased risk of complications at the time of delivery. Pathology
A single point measurement of SFH is at best an estimate of fetal size, rather than growth. Detection of aberrant growth requires serial measurements. The performance of SFH as a screening test for small babies has been reported to vary from 26% to 76%. This large variation in detection rate of IUGR in different studies may partly be explained by the ‘Hawthorne effect’: clinicians take extra care when they know they are being studied.
SFH measurements should not be seen as an isolated technique, but as part of an overall clinical assessment of the fetomaternal wellbeing.
Whereas the American College of Obstetricians and Gynecologists recommend repeated SFH measurements and ultrasound assessment of fetal growth in the presence of serial discrepancies, the Cochrane collaboration does not. The Cochrane conclusion is based on one randomized Danish trial, showing no difference in perinatal morbidity or mortality between intervention and control groups. SFH measurement
Technique
To measure the SFH correctly a non-elastic measuring tape should be used. The pregnant woman should be semirecumbent, knees flexed, with an empty bladder, enough of the pregnant abdomen exposed to identify anatomical landmarks, and the uterus non-contracting. The uterine fundus should be identified with two hands on the abdomen. The tape-measure should be held with the centimetre marks on the underside to reduce bias.
There are several techniques described for obtaining the measurement. Westin, in the original SFH paper from 1977, describing what he called a ‘gravidogram’, suggested fixating the tape-measure at the symphysis pubis and identifying the fundus with the other hand, fingers extended, and the measuring tape sliding between the fingers of the fundal hand. The second method (Fig. 7.26.1), recommended by Gardosi, appears to be more common in UK practice today. The tape measure is fixed at the fundus with one hand and allowing the symphyseal hand to take the measurement. The tape measure should stay in contact with the skin along the abdomen, and the measurement should be obtained along the longitudinal axis of the uterus, without correction to the abdominal midline, if the highest fundal point is not in the midline.
Clinician bias
It is difficult and not practical to be blinded to the gestational length of the pregnant women we see in our antenatal clinics, and it is well known that awareness of gestational length at the time of measurement introduces measurement error. A variation in clinician ability to correctly identify the uterine fundus has been reported. There is also a good evidence of an interobserver variation, which, however, appears to be improving with experience and training. The consensus is that fundal height cannot reliably be measured by different observers in the same pregnancy with sufficient agreement to separate small and normal or large fundal heights. This severely limits its clinical usefulness as a screening tool for both IUGR and macrosomia.

Fig. 7.26.1 (a–c) Correct technique for measuring symphyseal fundal height, courtesy of Professor Jason Gardosi, Gestation network 2010 www.gestation.net.
Other factors
Other fetomaternal factors that may cause variation in SHF measurement beyond the size of the fetus are
• amount of amniotic fluid
• size of placenta
• fetal presentation
• amount of abdominal fat
• thickness of myometrium
• presence of large fibroids
• relationship of uterus to the bony pelvis
• full bladder
• multifetal pregnancy
• multiparity. Clinical management
A normal growth rate of the SFH of 1 cm per week is often assumed, based on existing SFH growth charts. When the SFH measurement appears to deviate from the standard, whether the standard is a fundal height growth curve or a simple gestational weeks ± 2-cm rule (derived from observations that most pregnancies stay within this range throughout pregnancy), the recommended practice is repeat measurement by the same practitioner 1 week later. If at that stage the measurement is still the same, or smaller, referral to ultrasound for growth scan is recommended.
Similarly two consecutive measurements by the same clinician exceeding the given standard should raise the suspicion of macrosomia, and warrant investigations for macrosomia and a diabetic pregnancy. Serial ultrasound of the macrosomic fetus may not be necessary in the absence of other pathology, but an assessment of the best mode of delivery should occur at, or around term.
Previous pregnancy records often provide useful information. An otherwise low-risk woman, with what is perceived as insufficient SFH growth, which has had similar SFH growth patterns in previous pregnancies, and given birth to normal size offspring, would not need serial ultrasounds, provided that no other risk factors are identified. Variation in SFH in different ethnic groups
The assumption that fundal height curves could be a useful adjunct to antenatal care in resource poor settings has been tested. In a small prospective study in India, a high sensitivity and specificity for both small for gestational age (SGA) and macrosomia as found using locally generated SFH curve. The authors noticed a difference of 3–4 cm compared with ‘Western’ fundal height curves and a corresponding birthweight difference of 500–600 g. In a larger study of Mozambican pregnant women with ultrasound-dated singleton pregnancies, locally generated charts were 0–3 cm lower in measurements at the same gestational interval as other (presumably western) charts.
It is thus likely that there are different SFH growth curves in different ethnic populations. Locally derived SFH charts will therefore perform better than more generic charts in terms of detecting abnormal growth. SFH measurements will only perform as well as the chart they are plotted on. SFH measurement in obese women
Owing to an increasing prevalence of obesity in women of reproductive age in the Western world, the validity of SFH in obese women needs particular consideration. There is evidence that tape measurement bias increases with increasing BMI. Thus we cannot, in our clinical practice, assume that fundal height measurement have an equal validity, and will perform equally well in obese women as it has been shown to do in studies of normal weight pregnant women. On the other hand, if we do not assess SFH, considering it less useful, then ultrasound assessment of fetal growth becomes the default screening tool, and the advantage of ultrasound (often with limited vision in women with increased BMI and therefore a poorer quality assessment) in screening for fetal growth aberrations in obese women has yet to been demonstrated.
In the absence of alternative strategies, however, a low threshold for ultrasound referral for fetal growth assessment in obese women is recommended. Customised fundal height measurements
There is thus good evidence for a significant maternal variation in fundal height and a given gestation. The often-used rule of gestational length in weeks ±2 cm has less value in a multiethnic setting, or, as we have seen, in a setting with an increasingly obese pregnant population. Serial measurements of fundal height can, however, be plotted on a ‘customized’ chart, controlling for physiological variables such as maternal height, weight, parity, and ethnic group, and has been shown to lead to increased antenatal detection of small and large babies. Although in a small randomized study the ‘customized’ chart detected 48% (compared with 29% in the control group, OR 2.2) of all SGA babies it is still disappointing to note that half the SGA infants were undiagnosed in pregnancy. However, it is reasonable to assume that in modern UK obstetric practice, with a multiethnic pregnant population and increasing contribution of obese pregnant women, the customized chart will perform better than a more generic one. Assessment of fetal growth in the future
In current UK and European antenatal care working practice, a large proportion of pregnant women see more than one practitioner during their antenatal care, with ensuing implications for the quality of SFH measurements. The default alternative to SFH is, as we have seen, screening for growth abnormalities by serial ultrasound scans, which is expensive, time-consuming and generates false-positive results. The randomized studies of detection rates, costs, and effects of screening for abnormal growth by SFH measurement compared with ultrasound have not been done.
Self-measurement by pregnant women has been suggested as a possible future. A UK blinded pilot study recorded positive results of training in SFH measurement of pregnant women themselves. The study population performed as well as midwives in repeated, weekly measurements. Self-assessment by, we assume, interested and compliant pregnant women, would certainly address the interobserver variation problems of SFH measurement, and is simple, non-invasive, inexpensive, and well in agreement with the current ethos of patient involvement in the care they receive. SFH measurement as screening for IUGR and macrosomia should not been discarded until the comparative trial with ultrasound, using the right charts and the right techniques, has been undertaken, but in our current clinical practice it appears increasingly difficult to use SFH as a discriminating screening tool for fetal growth abnormalities. Further reading
American College of Obstetricians and Gynecologists. Intrauterine growth restriction. ACOG Practise Bulletin 12. Washington: ACOG 2000.
Bailey SM, Sarmandal P, Grant JM. A comparison of three methods of assessing interobserver variation applied to measurement of the symphysis-fundal height. Br J Obstet Gynecol 1989;96:1266–71.
Boulos AN, Griffiths M, Allott H, et al. Trial of self- administered antenatal care: maternal symphysis fundal height measurements. J Obstet Gynaecol 1999; 19: 623.
Challis K, Osman NB, Nystrom L, et al. Symphysis-fundal height growth chart of an obstetric cohort of 817 Mozambican women with ultrasound dated singleton pregnancies. Trop Med Int Health 2002;7:678–84.
Engstrom JL, McFarlin BL, Sampson MB. Fundal height measurement Part 4-Accuracy of Clinicians’ identification of the uterine fundus during pregnancy J Nurse Midwifery 1993;38:318–23.
Engstrom JL, Piscioneri LA, Low LK, et al. Fundal height measurement. Part 3: The effect of maternal position on fundal height measurements. J Nurse Midwifery 1993;38:23–7.
Gardosi J, Francis A. Controlled trial of fundal height measurement plotted on customised antenatal growth charts. Br J Obstet Gynecol 1999; 106; 309–17.
Grover V, Usha R, Kalra S, et al. Altered fetal growth. Antenatal diagnosis by symphysis-fundal height in India and comparison with western charts. Int J Gynaecol Obstet 1991;35:231–4.
Hepburn M, Rosenberg K. An audit of the detection and management of small-for-gestational-age babies. Br J Obstet Gynaecol 1986;93:212–16.
Jelks A, Cifuentes R, Ross MG. Clinician bias in fundal height measurement. Obstet Gynecol 2007;110:892–9.
Lindhard A, Nielsen PV, Mouritzen LA, et al. The implications of introducing the symphyseal-fundal height-measurement. A prospective randomised controlled trial. Br J Obstet Gynecol 1990;97:675–80.
Neilson JP. Symphysis-fundal height measurement in pregnancy. Cochrane Database Syst Rev 1998; 1:CD000944.
Parson HM. What happened at Hawthorne? Science 1974;198:922–32.
Pearce JM, Campbell S. A comparison of symphysis-fundal height and ultrasound as screening tests for light-for-gestational-age infants. Br J Obstet Gynecol 1987;94:100–4.
Petzold M, Sonesson C, Bergman E, et al. Surveillance in longitudinal models: detection of intrauterine growth restriction. Biometrics 2004;60:1025–33.
Rogers MS, Chan E, Ho A. Fundal height: does prior knowledge of gestational age influence the measurement? J Obstet Gynecol 1992;12:4–5.
Rogers MS, Needham PG. Evaluation of fundal height measurement in antenatal care. Aust NZ J Obstet Gynecol, 1985;25:87–90.
Steingrimsdottir T, Cnattingius S, Lindmark G. Symphysis-fundus height: construction of a new Swedish reference curve, based on ultrasonographically dated pregnancies. Acta Obstet Gynecol Scan 1995;74:346–51.
Westin B. Gravidogram and fetal growth. Acta Obstet Gynecol Scand 1977;56:273–82. Internet resources
West Midlands Perinatal Institute: www.pi.nhs.uk/growth Patient resources
Generating your own customised fetal growth and SFH chart: www.gestation.net