39 Evaluation of Amenorrhea
Irene Woo
Kyle J. Tobler
Amenorrhea is the absence of menses. It is physiologic during pregnancy, lactation, and menopause. The lack of regular, spontaneous menses for any other reason after the expected age of menarche is pathologic.
• Primary amenorrhea: No menses by age 14 years in the absence of secondary sexual development or no menstruation by age 16 years with the presence of secondary sexual characteristics.
• Secondary amenorrhea: The absence of menses in a previously menstruating woman. It is also defined as the lack of menses for 6 months or for three menstrual cycles in women that have experienced menarche. Evaluation, however, need not be deferred solely to conform to these definitions.
• World Health Organization (WHO) amenorrhea groups
• WHO group I (hypogonadotropic hypoestrogenic) has no endogenous estrogen production, normal or low follicle-stimulating hormone (FSH) levels, normal prolactin (PRL) levels, and no lesion in the hypothalamus or pituitary.
• WHO group II (normogonadotropic normoestrogenic) has endogenous estrogen production and normal levels of FSH and PRL.
• WHO group III (hypergonadotropic hypoestrogenic) has elevated FSH levels and low to absent estrogen, indicative of premature ovarian failure (POF).
MENSTRUAL PHYSIOLOGY
• Spontaneous, cyclic menstruation requires an intact and functional hypothalamicpituitary-ovarian axis (HPOA), endometrium, and genital outflow tract. Abnormalities in any of these structures may result in amenorrhea.
Normal Physiology of the Hypothalamic-Pituitary-Ovarian Axis and Menstruation
• Hypothalamus (arcuate nucleus) secretes gonadotropin-releasing hormone (GnRH) in pulses at specific frequencies and amplitudes into the portal circulation.
• GnRH pulses stimulate gonadotrophs in the anterior pituitary to synthesize, store, and secrete gonadotropic hormones FSH and luteinizing hormone (LH) to the systemic circulation.
• FSH stimulates ovarian follicle development and estradiol (E2) secretion.
• E2 provides inhibitory feedback on the hypothalamus and pituitary, decreasing FSH release. E2 also stimulates proliferation of the endometrium.
• Follicular growth continues until the threshold level of systemic E2 is surpassed, shifting to positive feedback, which triggers the LH surge.
• LH surge causes the developing oocyte within the follicle to resume meiosis and ovulate.
• Following ovulation, the follicle becomes a corpus luteum, rapidly shifting from primarily E2 production to progesterone production.
P.514
• Progesterone decidualizes the endometrium in preparation for embryo implantation.
• If pregnancy occurs, human chorionic gonadotropin (hCG) secreted from the Syncytiotrophoblast supports the corpus luteum and continued progesterone release.
• If pregnancy does not occur, the corpus luteum will regress, ceasing to produce progesterone.
• Progesterone withdrawal causes the endometrium to slough, resulting in the menstrual effluent.
• The HPOA and endometrium demonstrate a finely orchestrated system that must be intact at all steps for a normal menstrual cycle to occur (Fig. 39-1).
EVALUATION OF AMENORRHEA
When to Evaluate for Amenorrhea
• Rule out pregnancy, as both primary and secondary amenorrhea require an immediate evaluation for pregnancy.
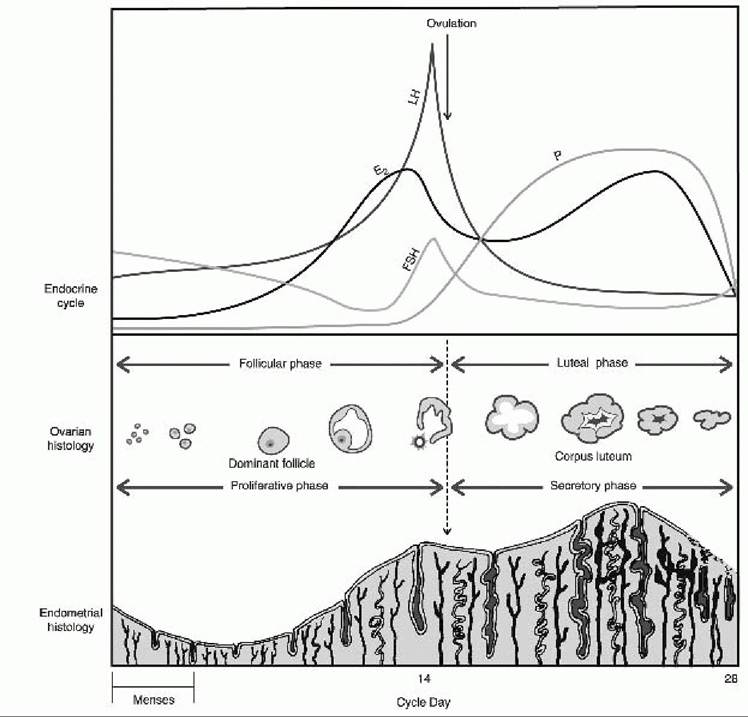
Figure 39-1. The normal menstrual cycle. Changes in serum hormones, ovarian follicle, and endometrial thickness during a 28-day menstrual cycle. Menses occur during the first few days of the cycle. E2, estradiol; FSH, follicle-stimulating hormone; LH, luteinizing hormone; P, progesterone. (From Berek J, ed. Berek and Novak’s Gynecology, 14th ed. Philadelphia, PA Lippincott Williams & Wilkins, 2007.)
P.515
• Use clinical judgment. The aforementioned listed timeline defining amenorrhea does not need to be met prior to initiating an evaluation.
• Do not overlook gross evidence of a disease process: T urner syndrome, frank virilization, obstructed vagina, or other evidences of a disease process.
• Use a systematic approach, evaluating each critical component of menstruation: hypothalamus, pituitary, ovaries, uterus, and genital outflow tract.
Important History for Amenorrhea
• Present illness: Presence of cyclic pelvic or abdominal pain, headache, visual changes, seizure, hot flushes, hot or cold temperature intolerance, vaginal dryness, urinary issues, hirsutism, virilization, galactorrhea, severe physical or emotional stress, changes in weight, diet, athletic training, or trauma
• Past medical history: General health; chronic illnesses (especially autoimmune and thyroid disease); birth defects; all current and recently discontinued medications or supplements; contraception history (especially the use of depot medroxyprogesterone acetate); history of pelvic infection, complications with prior pregnancies or abortions, and any instrumentation of the uterus; and abdominal or pelvic surgeries. Most recent pregnancy and delivery and lactation history can be significant, as can a personal history of cancer treatment involving radiation therapy and/or chemotherapy.
• Development: Age of thelarche, pubarche, and menarche, whether menarche was spontaneous or induced, and cycle regularity
• Social: Severe physical or emotional stress, changes in weight or diet, and athletic training
• Family history: History of late pubertal development, early menopause, mental retardation, or short stature
Important Physical Examination for Amenorrhea
• Height, weight, body mass index, waist-to-hip ratio if obese, blood pressure, and pulse
• General body habitus, looking for disease stigmata of Turner syndrome, Cushing syndrome, and thyroid disease. Also gross malnutrition or obesity.
• Vision changes or peripheral loss of vision
• Mouth and teeth for tooth enamel erosion
• Skin evaluated for hyperpigmentation, acanthosis nigricans, abdominal striae, acne, hirsutism, and balding
• Thyroid gland palpated for size, shape, and nodules
• Breast development (T anner stage), galactorrhea, or other breast discharge
• Abdominal exam for masses, fat distribution, hirsutism, and aforementioned listed skin changes
• External genitalia examined for hair distribution and virilization (clitoromegaly), imperforate hymen, or labial fusion
• Internal genitalia examined for transverse vaginal septum, lateral vaginal obstruction, estrogenized vaginal mucosa, and the presence of a cervix with visible patent external cervical os
• Rectal exam to evaluate the extent of hematocolpos and presence of uterus beyond a vaginal obstruction or absent vaginal orifice.
Rectal exam can also assist in evaluating a patient with an intact hymen or infantile vaginal orifice.Laboratory Evaluation of Amenorrhea
• Important for laboratory evaluation to be guided by the aforementioned presenting history and physical examination
hCG to evaluate for pregnancy
P.516
• FSH, E2, thyroid-stimulating hormone (TSH), and PRL
• 17-Hydroxyprogesterone, testosterone, and dehydroepiandrosterone sulfate (DHEAS) for patients with virilization, hirsutism, or androgen excess
• Testosterone if concern for complete androgen insensitivity
• Karyotype if concern for genitourinary abnormalities, suspicion for gonadal dysgenesis, or complete androgen insensitivity. Also consider if other nonrelated physical malformations are present.
Imaging Evaluation of Amenorrhea
• Pelvic ultrasound for both primary and secondary amenorrhea
• Hysterosalpingogram (HSG) or sonohysterography particularly for secondary amenorrhea and suspicion for Asherman syndrome
Follow-Up Laboratory and Imaging Studies for Initial Evaluation
• Fragile X (FMR1) premutation for patients with POF
• Antiadrenal antibodies and antithyroid antibodies (antiperoxidase and antithyroglobulin) for patients with POF
• Karyotype for patients younger than 30 years with POF
• Cortisol levels (24-hour urinary free cortisol, late-night salivary cortisol, dexamethasone suppression testing) for patients with suspected Cushing syndrome, also considered in patients evaluated for polycystic ovarian syndrome (PCOS) and/or evidence of hyperandrogenism
• Insulin-like growth factor 1 (IGF-1), free T4, and morning cortisol level for patients with a pituitary lesion identified by magnetic resonance imaging (MRI)
• Adrenocorticotropic hormone (ACTH) stimulation test for patients with elevated 17-hydroxyprogesterone
• MRI of the pituitary for hyperprolactinemia or hypogonadotropic hypogonadism which has no other identifiable etiology (severe physical and emotional stress, malnutrition, medications, hypothyroidism)
• MRI of pelvis.
Obtain when genitourinary abnormalities are not well characterized or for surgical planning. Particularly useful when evaluating for imperforate hymen versus transverse vaginal septum, obstructed hemivagina, and noncommunicating or hypoplastic uterine horn.• Renal ultrasound and radiographs (computed tomography [CT] or x-ray) of spine for patients with mullerian dysgenesis
• Endometrial biopsy (suspicion of genital tuberculosis or schistosomiasis)
Progesterone Withdrawal for Evaluation of Amenorrhea
• Progestin challenge: 5 to 10 mg of medroxyprogesterone (Provera) for 5 to 7 days. Positive response is withdrawal bleed within 2 to 7 days after discontinuation of Provera.
• Approximately 20% of patients with POF, hypothalamic amenorrhea, and hyperprolactinemia experience withdrawal flow depending on the degree of hypoestrogenism.
• Failure to withdraw after sequential estrogen then estrogen/progestin is supportive of Asherman syndrome or cervical stenosis, but these conditions are rarely seen in the absence of a previous surgical procedure and the amenorrhea can be temporally related to the procedure.
• Consider use of serum E2 level rather than use of progesterone withdrawal to determine status of estrogen.
• Progesterone-induced withdrawal bleed is indicated as a treatment for amenorrhea and a thickened endometrium on ultrasound.
P.517
TABLE 39-1 Differential Diagnosis of Primary Amenorrhea
| Breast Development Present | Breast Development Absent | |
| Uterus present | Consider secondary amenorrhea differential Hypothalamic cause Pituitary cause Ovarian cause Uterine cause | Gonadal dysgenesis 45, X 46, X; abnormal X Mosaic X 46,XX or 46,XY: pure gonadal dysgenesis 17-hydroxylase deficiency with 46,XX Galactosemia |
| Hypothalamic or pituitary failure | ||
| Kallmann syndrome CNS congenital defect Hypothalamic-pituitary tumors CNS infection Physiologic delay | ||
| Uterus absent | Mullerian agenesis Androgen insensitivity syndrome | 17,20-desmolase deficiency (46,XY) Agonadism 17-hydroxylase deficiency (46,XY) |
CNS, central nervous system.
Differential Diagnosis for Primary Amenorrhea
• History and physical examination to evaluate for genital outflow obstruction
• Keep pregnancy in differential, although less likely.
• Keep the most common causes high on differential, (gonadal dysgenesis, mullerian anomalies/dysgenesis, and complete androgen insensitivity).
• To develop a differential diagnosis, categorize the patients into four categories based on the presence or absence of a uterus and the presence or absence of breast development (indicative of estrogen) (T able 39-1).
• Uterus present and breasts absent likely represents gonadal dysgenesis, hypothalamic failure, or pituitary
failure.
• Uterus absent and breasts present likely represents androgen insensitivity or congenital absence of the uterus.
• Both uterus and breasts absent likely represents failure of steroidogenesis to produce sex hormones including 17- or 20-desmolase deficiency, 17α-hydroxylase deficiency, or agonadism. Frequently have 46,XY karyotype combined with gonadal failure.
• Both uterus and breast development present likely represents pituitary etiology (hyperprolactinemia) or another subcategory that also underlies secondary amenorrhea.
Differential Diagnosis for Secondary Amenorrhea
• Always keep pregnancy high on the differential diagnosis (Table 39-2).
• Physiologic explanations include pregnancy, menopause, and postpartum lactation.
P.518
TABLE 39-2 Pathologic Causes of Secondary Amenorrhea
| Etiology | Causal Factor |
Reproductive tract
| Cervical stenosis | Surgical procedure (i.e., LEEP, CKC) |
| Asherman syndrome | Endometrial scarring |
Ovarian
| Premature ovarian failure | Idiopathic, chromosomal abnormality, autoimmune disease, infection |
| Polycystic ovary syndrome | Inappropriate gonadotropin secretion, insulin resistance |
Pituitary
| Hyperprolactinemia | Lactotroph hyperplasia ± prolactinoma, drugs |
| Pituitary adenomas | Thyrotroph, corticotroph, or other hyperplasia |
| Sheehan syndrome | Postpartum hemorrhage |
CNS
Hypothalamic amenorrhea
Stress, eating disorders, weight loss, excessive
exercise
Brain injury Interruption of HPOA
Inflammatory or infiltrative process Interruption of HPOA
Other endocrinopathies
Hypothyroidism, Cushing syndrome, late-onset
adrenal hyperplasia
LEEP, loop electrosurgical excision procedure; CKC, cold knife conization; CNS, central nervous system; HPOA, hypothalamic-pituitary-ovarian axis.
• If onset is related to previous pregnancy, abortion, or other surgical procedure, consider cervical stenosis or Asherman syndrome. Further evaluation with HSG, hysteroscopy, or sonohysterogram.
• Mildly elevated PRL: Repeat in the morning (patient needs to refrain from breast stimulation, intercourse, or exercise prior to test). Also, verify normal TSH to rule out hypothyroidism as etiology of hyperprolactinemia. MRI will confirm presence of a pituitary lesion.
• Normal E2 and FSH levels: Likely anovulation, consider further evaluation for PCOS.
• Low E2 and low FSH levels: Consider central nervous system (CNS) lesion or hypothalamic-pituitary failure and further evaluation with MRI.
• Low E2 and elevated FSH levels: Consider POF and gonadal dysgenesis.
• Elevated TSH: occult or subclinical hypothyroidism
• Elevated DHEAS: Rule out adrenal tumor with CT scan.
• Elevated 17-hydroxyprogesterone: Consider late-onset congenital adrenal hyperplasia and confirm with ACTH stimulation test.
P.519
• Evidence of androgen excess: with a normal E2, FSH, PRL, TSH, 17-hydroxyprogesterone and DHEAS, should consider PCOS. May see polycystic ovaries on pelvic sonogram; however, it is not required for diagnosis.
• Signs or symptoms of Cushing syndrome: Screen with late-night salivary cortisol (easiest), 24-hour urinary free cortisol, 1-mg overnight dexamethasone suppression, or 2-day low-dose dexamethasone suppression screening tests.
ETIOLOGIES OF AMENORRHEA—SYSTEMATIC EVALUATION
Genital Outflow Tract and Uterine Abnormalities Resulting in Amenorrhea
• Imperforate hymen and transverse vaginal septum are outflow tract malformations that typically present with acute cyclic pelvic or abdominal pain in a patient soon after the age of expected menarche. They will often have age-appropriate secondary development. Examination of an imperforate hymen reveals no obvious vaginal orifice and often a bulging, thin perineal membrane. In a patient with a transverse septum, physical exam will reveal a normal vaginal orifice but no visible cervix. Imperforate hymen should distend with Valsalva maneuver. In some cases, an MRI may be required to distinguish an imperforate hymen from a transverse septum.
• Mullerian agenesis and hypoplasia, also known as Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome, is a relatively common cause of primary amenorrhea. The incidence ranges from 1:4,000 to 1:10,000. Subjects with MRKH commonly present in their late teens with normally developing breasts, pubic hair, and external genitalia, as the presence and function of the ovaries are normal. Depending on the location of the mullerian agenesis, the patient can present with no vagina, a portion of a vagina as well as complete uterine agenesis, or a portion of the uterus. Amenorrhea is generally the only complaint, although 2% to 7% may have rudimentary mullerian structures with functioning endometrium, resulting in cyclic pain. MRI of the pelvis can assist with classifying the anomalies and surgical planning, if required. In addition, imaging of the urinary tract should be performed in all patients with mullerian abnormalities, as approximately 30% have renal anomalies. Skeletal abnormalities are also commonly associated with MRKH. Vaginal dilator therapy or surgical construction of a neovagina can usually create a functional vagina.
• Complete androgen insensitivity syndrome (CAIS), previously known as testicular feminization, is an X- linked, recessive disorder that occurs in 46,XY individuals and results in phenotypic women. Testes are present and secrete normal male levels of anti-mullerian hormone (AMH) and testosterone. AMH results in regression of mullerian structures. Masculinization fails to occur because of an androgen receptor defect. Like MRKH, patients with CAIS typically present in the later teens with normal development of breasts with primary amenorrhea. Physical examination generally demonstrates normal external genitalia, a shortened or absent vagina, and no cervix or uterus. Also, physical exam can often differentiate the two conditions because pubic and axillary hair is sparse in CAIS, and testes may be palpable in the inguinal region. The diagnosis of CAIS is confirmed by documenting serum testosterone in the normal male range and a 46,XY karyotype. The incidence of gonadal neoplasia is 52%, and the incidence of gonadal malignancy is 22% in CAIS. Under these circumstances, gonadectomy must be performed. Because malignancy rarely occurs before age 20 years, deferring surgery until after pubertal maturation and epiphyseal closure have occurred is preferable. The gynecologist should refer to the removed tissue as
P.520 gonad rather than testis in initial discussions with the patient. Counseling is often indicated. Vaginal dilator therapy can usually create a functional vagina.
• Asherman syndrome is the most common cause of secondary amenorrhea and accounts for 7% of patients presenting with secondary amenorrhea. Asherman syndrome (i.e., intrauterine synechiae) is most commonly associated with aggressive postpartum curettage or abortion. Other risk factors include uterine or cervical surgeries, such as cesarean section, septoplasty, myomectomy, and cone biopsy procedures. Infectious causes include tuberculosis, schistosomiasis, infection associated with intrauterine devices (IUDs), and other severe pelvic infections. Diagnosis can be confirmed with HSG, sonohysterogram, or hysteroscopy. Treatment requires hysteroscopic lysis of intrauterine adhesions and placement of intrauterine stent.
• Cervical stenosis can be the result of congenital defects or acquired following cervical conization or loop electrosurgical excision and dilation and curettage. If cervical stenosis is the underlying etiology of secondary amenorrhea, hematometra and an enlarged uterus should be detected by physical exam and confirmed with ultrasound. Treatment includes serial dilation of the cervix.
Ovarian Abnormalities Resulting in Amenorrhea (Hypergonadotropic Hypogonadism)
• Primary dysfunction at the level of the ovary. The ovaries no longer respond to gonadotropin stimulation limiting follicular development and production of E2.
• Gonadal dysgenesis is the most common cause of primary amenorrhea, accounting for 43% of such cases. Peripheral blood karyotype aids in diagnosis. Although T urner syndrome is the most frequent cause of gonadal dysgenesis, any condition resulting in depletion of germ cells can cause gonadal dysgenesis and replacement of the gonads with fibrous streaks.
• Turner syndrome (TS) classically results from aneuploidy involving the X chromosome. Approximately, 60% of TS patients are 45,X and the other 40% include karyotype abnormalities such as 45,X/46,XX mosaics; 46,XXqi isochromosome; and 46,XXp short arm deletion. Internal and external genitalia develop normally for females. The cohort of primordial follicles undergoes accelerated atresia so that oocytes are depleted prior to the onset of puberty. A lack of gonadal E2 production results in a failure of breast development and other secondary sexual characteristics.
î Patients with TS exhibit several cardinal features including webbed neck, shieldshaped chest, short stature, and sexual infantilism. Typically, these patients are identified in the pediatric population due to short stature, prior to noting primary amenorrhea. Some TS patients, especially those with mosaic karyotypes, can undergo spontaneous puberty and conception (16% and 3.6% of cases, respectively).
• Mosaicism involving partial deletions or rearrangements of one X chromosome can cause a wide range of gonadal dysfunctions ranging from gonadal dysgenesis to POF. Determining whether a Y chromosome is present in a mosaic is important because the presence of the SRY portion of the Y chromosome predisposes to tumor formation. Presence of a Y chromosome requires gonadectomy or removal of the gonadal streaks.
• Pure gonadal dysgenesis is a term used to describe 46,XX or XY individuals who experience dysgenesis of germinal tissue early in embryonic development. Such dysgenesis likely results from genetic, environmental, or infectious insults, although a specific cause is rarely identified. All subjects are phenotypic women of normal height who fail to undergo puberty. Patients with 46,XY gonadal dysgenesis, also known as Swyer syndrome, require removal of their gonadal streaks to prevent malignant transformation.
P.521
• CYP17 deficiency is a rare disorder that can affect 46,XY or XX individuals. The lack of 17α-hydroxylase and 17,20-lyase activities results in both gonadal and adrenal insufficiencies. Patients with an XY karyotype are phenotypic women (due to lack of androgen production) but also lack a uterus because AMH was secreted in early fetal life. Subjects usually present at the time of puberty with hypertension (due to excess mineralocorticoid production), hypokalemia, and hypergonadotropic hypogonadism. CYP17 deficiency is an autosomal recessive disorder.
• LH and FSH receptor mutations have also been identified preventing the ovaries from responding to gonadotropin stimulation and resulting in POF. They can present with varying levels of secondary sexual development and likely primary amenorrhea. However, these conditions are very rare.
• POF can manifest as primary or secondary amenorrhea. For patients who previously menstruated, POF is defined as amenorrhea associated with a depletion of oocytes and cessation of menses before age 40 years. In those with primary amenorrhea, approximately 50% will have an abnormal karyotype. The various possible etiologies for POF associated with secondary amenorrhea are listed in the following text; however, up to 90% of patients with POF remain unexplained following evaluation.
• X chromosome abnormalities, such as short or long arm deletions or mosaicism, not severe enough to cause primary gonadal dysgenesis, may present as POF.
• Spontaneous POF is not induced by chemotherapy, radiation, or surgery. The majority of cases are idiopathic; 6% have premutations in the gene responsible for fragile X syndrome (FMR1); and 4% have steroidogenic cell autoimmunity, placing them at risk for adrenal insufficiency. Because 14% of patients with familial POF and 2% of isolated POF will have the FMR1 premutation, it is important to evaluate for the FMR1 gene premutation by obtaining a family history of POF, fragile X, unexplained mental retardation, tremor/ataxia syndrome, and/or any developmental delay in children. In addition, because up to 20% of patients with POF develop autoimmune hypothyroidism, they should undergo adrenal and thyroid antibody testing. Those younger than age 30 years with POF should also have karyotyping performed, as 13% will show some chromosomal abnormalities. Inclusion of any Y chromosomal material is an indication for gonadectomy.
• Iatrogenic POF can be the result of follicular depletion by radiation, chemotherapy (especially with alkylating agents), or surgical manipulation or removal of ovarian tissue. Prior to undergoing radiation or chemotherapy, measures can be taken to decrease exposure to or mitigate damage. Prior to radiation therapy, oophoropexy can position ovaries outside the radiation field. Prior to and throughout chemotherapy treatment for malignancy or severe autoimmune diseases, GnRH agonists or antagonists can potentially provide protection, although the efficacy of these treatments is still debated. Additionally, many centers offer ovarian tissue cryopreservation; however, the optimal strategies and protocols are still under investigation.
• Treatment of POF involves estrogen replacement and should be initiated in essentially all patients to prevent the premature onset of osteopenia and osteoporosis. In addition, these women are at high risk for early-onset cardiovascular disease, genitourinary atrophy, vasomotor symptoms, sleep disturbance, and vaginal dryness. Often, POF patients require twice as much estrogen as compared to postmenopausal women to alleviate symptoms. This can be accomplished with use of oral contraceptive pills or higher doses of traditionally used hormone replacement therapy regimens (e.g., micronized E2 1 to 2 mg daily or conjugated equine estrogens 0.625 to 1.25 mg daily) or transdermal treatment regimens
P.522 (0.1 mg/24 hours). In patients with short stature or open epiphyseal plates, lower doses of estrogen should be used to avoid premature closure. If the uterus is intact, adjunct cyclic treatment with progestins is required to prevent endometrial hyperplasia.
• Spontaneous pregnancy following POF is possible, although unlikely (~5%). T reatment of infertility classically requires oocyte donation; however, in some cases, high doses of gonadotropins can achieve follicular development. Lastly, POF can be associated with psychological distress and appropriate counseling and emotional support should be initiated.
Hypothalamic Dysfunction Resulting in Amenorrhea (Hypogonadotropic Hypogonadism)
• Underlying etiology is a decrease in GnRH release and stimulation of the pituitary to release gonadotropins resulting in failure of folliculogenesis and production of E2.
• The term hypothalamic amenorrhea applies to conditions in which GnRH secretion is diminished in the absence of any organic pathology.
• Physical or psychological stress, anorexia nervosa, exercise, and weight loss can contribute to dysfunctional hypothalamic GnRH secretion. Affected women are frequently underweight, >10% below ideal body weight, and/or engage in regular strenuous exercise.
• Kallmann syndrome is an inherited X-linked disorder resulting from a genetic mutation that causes failure of olfactory and GnRH neuronal migration from the olfactory placode. The resultant hypogonadotropic hypogonadism is due to the absence of GnRH pulses to stimulate gonadotropin release from the pituitary. This syndrome is characterized by primary amenorrhea, absent breast development, presence of cervix and
uterus, and anosmia.
• Congenital GnRH deficiency is a genetic condition resulting in the absence of functional hypothalamic neurons. Unlike Kallmann syndrome, it is not associated with anosmia.
• GnRH receptor mutations inhibit signaling of GnRH to release gonadotropins from the anterior pituitary. Patients affected have a broad range of phenotypes, depending on the particular mutation.
• Other CNS pathologies, such as hypothalamic neoplasms, trauma, hemorrhage, or cranial irradiation, can interrupt the function of the HPOA. Craniopharyngioma is the most common CNS neoplasm causing delayed puberty. An MRI should be ordered for any patient with hypogonadotropic amenorrhea when no obvious external cause is present.
• Chronic debilitating disease can also lead to hypogonadotropic amenorrhea as a result of alterations in GnRH pulsatility. This has been observed in renal disease, liver disease, malignancy, and HIV. However, virtually any serious chronic illness can undermine the HPOA.
• Treatment involves correcting the underlying causative behavior if identified. The primary treatment is estrogen/progestin replacement as described in the POF treatment section.
Pituitary Disorders Resulting in Amenorrhea
• Pituitary lesions can present with amenorrhea and low or normal levels of gonadotropins. The most common pituitary lesion is a prolactinoma, but nonfunctioning adenomas, adenomas that secrete other pituitary hormones, or empty sella syndrome may also be present.
P.523
• Hyperprolactinemia accounts for 14% of secondary amenorrhea and a small portion of primary amenorrhea. Pregnancy and breast-feeding are physiologic causes of hyperprolactinemia. Medications that can cause hyperprolactinemia include most antipsychotics and antidepressants, H2 receptor blockers, methyldopa, verapamil, reserpine, and metoclopramide. Other medical causes that must be evaluated include hypothyroidism and renal failure. However, by far the most common pathologic cause of hyperprolactinemia is a prolactinoma.
• Prolactinomas are classified as either microadenomas or macroadenomas (>10 mm). Macroadenomas may be associated with bitemporal hemianopsia and therefore, visual field defects should be evaluated for during physical examination.
• Excess PRL levels can cause negative feedback on hypothalamic GnRH secretion, thereby lowering gonadotropin release. In addition to hypogonadism, most women will experience oligo or amenorrhea and galactorrhea. Galactorrhea is secretion of a milky fluid, excluding breast-feeding. Discharge may be white/clear in color but also greenish or even bloody. Bloody discharge requires evaluation for an intraductal papilloma or cancer with mammography. In the absence of hyperprolactinemia, galactorrhea does not need further workup.
• Laboratory evaluation of serum PRL levels between 20 and 200 ng/mL are considered elevated. A mildly elevated serum PRL should be repeated in a fasting, nonstressed environment because PRL concentration can vary with time of day, level of stress, and other factors. Occasionally, macroadenomas can produce extremely high serum PRL levels (>1,000 ng/mL), but the laboratory value could be falsely low due to the hook effect. In this phenomenon, the substrate saturates both the capture and signal antibodies used in the laboratory assays resulting in inability of the two antibodies to bind. This results in only a modestly elevated level in the sample. If clinical suspicion is high for hyperprolactinemia but the laboratory value is inconsistent, the test should be repeated with the serum diluted. Women with pituitary macroadenomas
should also have additional evaluation including a serum free T4, IGF-1, and morning cortisol level.
• A confirmed elevation in PRL prompts imaging of the pituitary gland, usually by MRI. At least 30% to 40% of women with hyperprolactinemia have a pituitary adenoma. The incidence of malignancy in prolactinomas is very rare and resection is rarely required.
• T reatment of hyperprolactinemia is usually successful with dopamine agonist therapy (bromocriptine or cabergoline). See Chapter 13.
• Sheehan syndrome is a condition of pituitary necrosis and hypopituitarism following postpartum hemorrhage and hypotension. See Chapter 13.
• Infiltrative disease: most commonly caused by hemochromatosis, a disorder of excessive deposition of iron in liver, pancreas, anterior pituitary, and heart. Screen iron studies; fasting transferrin saturation >45% is indicative. Treat with phlebotomy and chelation therapy.
• Isolated gonadotropin (FSH/LH) deficiency is a rare condition usually associated with thalassemia major, retinitis pigmentosa, or prepubertal hypothyroidism.
Normogonadotropic Amenorrhea
• Heterogeneous group with normal levels of gonadotropins and E2. Patients have normal secondary sexual development. The underlying etiology of amenorrhea is chronic anovulation.
P.524
• PCOS is the most common cause of amenorrhea associated with hyperandrogenism. Patients generally have normal levels of gonadotropins and E2.
• National Institutes of Health and the Rotterdam Consensus Conference are the most commonly used diagnostic criteria for PCOS. See Chapter 41.
• After other causes of amenorrhea and hyperandrogenism have been excluded, evidence of chronic an- /oligoovulation, androgen excess, and/or polycystic ovaries on ultrasound generally establishes the diagnosis.
• Hyperandrogenism should be evaluated with serum testosterone and 17-hyroxyprogesterone and DHEAS levels should be assessed to exclude the late-onset congenital adrenal hyperplasia or the presence of an adrenal or other androgen-producing tumors.
• PCOS is associated with an increased risk for type 2 diabetes, insulin resistance, hypertension, lipid abnormalities, obesity, metabolic syndrome, and endometrial hyperplasia/cancer.
• Treatment includes weight reduction and inducing withdrawal bleed through cyclic progesterone or a combined hormonal contraceptive to decrease risk of unopposed estrogen stimulation of uterine lining, also identification and treatment of other underlying medical comorbidities (diabetes, obesity, hyperlipidemia, hirsutism).
• Late-onset congenital adrenal hyperplasia generally presents similarly to PCOS with amenorrhea and hyperandrogenism. Initial screening with 17-hydroxyprogesterone followed by ACTH stimulation test establishes the diagnosis. Most patients have an autosomal recessive disorder resulting in 21-hydroxylase deficiency. T reatment of amenorrhea involves glucocorticoid replacement and/or combined contraception. See Chapter 41 for a more complete discussion.
• Cushing syndrome is a clinical state resulting from prolonged, inappropriate hypercortisolism. Etiology includes pituitary tumor (Cushing disease), adrenal hypersecretion of cortisol, or iatrogenic (chronic steroid use). It is characterized by loss of normal hypothalamic-pituitary-adrenal feedback mechanisms and loss of the normal circadian rhythm of cortisol secretion. Screening tests include late-night salivary cortisol (evaluated diurnal variation), 24-hour urinary free cortisol (evaluates secretion), and dexamethasone suppression testing (evaluates impaired feedback).
• Hyperprolactinemia frequently presents with normal gonadotropin levels and normal to mildly depressed E2. See earlier discussion.
• Thyroid disease can present with normal levels of gonadotropins and amenorrhea. Classically, hypothyroidism accounts for 1% to 2% of primary and secondary amenorrhea. Hypothyroidism can lead to hyperprolactinemia. Thyrotropin-releasing hormone (TRH) stimulates the release of TSH and PRL from the anterior pituitary. Therefore, patients with poorly controlled hypothyroidism may also experience sequelae of hyperprolactinemia. Both PRL and TSH should be routinely evaluated as part of the evaluation for amenorrhea. An elevated TSH and low T4 confirm hypothyroidism. An elevated TSH and normal T4 is diagnostic for subclinical hypothyroidism. Both clinical and subclinical hypothyroidism should be treated.
T reatment should be initiated with 25 to 50 pg/day of levothyroxine followed by TSH assessment every 4 to 6 weeks until TSH levels normalize.
Menopause
• Menopause occurs secondary to a genetically programmed loss of ovarian follicles. The onset of menopause should be differentiated from POF based on the age of the
P.525 patient. It is defined as 12 months of amenorrhea after the final menstrual period. It reflects complete, or near complete, ovarian follicular depletion and the absence of ovarian E2 secretion. Mean age of menopause is 51 years with a range of 43 to 57 years in American women. It is characterized by elevated FSH and low E2. See Chapter 43.
SUGGESTED READINGS
American College of Obstetricians and Gynecologists. ACOG practice bulletin no. 108: polycystic ovary syndrome. Obstet Gynecol 2009;114:936-949.
Nelson LM, Covington SN, Rebar RW. An update: spontaneous premature ovarian failure is not an early menopause. Fertil Steril 2005;83:1327-1332.
Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea.
Fertil Steril 2008;90:S219-S225.
Rebar RW, Connolly HV. Clinical features of young women with hypergonadotropic amenorrhea. Fertil Steril 1990;53:804-810.
Reindollar RH, Byrd JR, McDonough PG. Delayed sexual development: a study of 252 patients. Am J Obstet Gynecol 1981;140:371-380.
Reindollar RH, Novak M, Tho SP, et al. Adult-onset amenorrhea: a study of 262 patients. Am J Obstet Gynecol 1986;155:531-543.
Speroff L, Fritz MA. Clinical Gynecologic Endocrinology and Infertility, 8th ed. Philadelphia, PA Lippincott Williams & Wilkins, 2011.
Zacur HA. Indications for surgery in the treatment of hyperprolactinemia. J Reprod Med 1999;44:1127-1131.