Ovary
Ovarian tumours
There are many types of ovarian tumours including benign, borderline, and malignant types. About two-thirds occur in women of reproductive age. Approximately 80% of ovarian tumours are benign.
Almost 90% of malignant and borderline tumours are diagnosed after the age of 40 years (1, 8).Ovarian tumours are classified by the cell type of origin. Most are common epithelial tumours (approximately 60%). Other important groups are germ cell tumours (30%), sex cord/s tromal tumours (8%), and tumours metastatic to the ovary. Common epithelial tumours account for about 90% of ovarian malignancies, high-grade serous adenocarcinoma being the most common (70%).
Ovarian cancer is the second most frequent gynaecological malignancy after endometrial cancer and carries a higher mortality rate than all other female genital cancers combined. As it is difficult to detect early in its evolution when it is still curable, over three- quarters of patients already have extraovarian tumour spread to the pelvis or abdomen at the time of diagnosis (1, 8).
Epithelial tumours
Tumours of common epithelial origin can be broadly classified, according to cell proliferation, degree of nuclear atypia, and presence or absence of stromal invasion: (1) benign, (2) of borderline malignancy, and (3) carcinoma.
Common epithelial neoplasms most commonly affect nulliparous women and occur least frequently in women in whom ovulation has been suppressed (e.g. by pregnancy or oral contraceptives). Whereas the lifetime risk for developing ovarian cancer in the general population is 1.6%, women with one first-degree relative with ovarian cancer have a 5% risk. Also, women with a family history of ovarian carcinoma are at greater risk for breast cancer and vice versa. Defects in repair genes implicated in hereditary breast cancers, BRCA1 and BRCA2, are incriminated in familial ovarian cancers as well.
As for endometrial carcinoma, women with hereditary non- polyposis colon cancer (HNPCC) are also at greater risk for ovarian cancer (1, 8).Epithelial ovarian tumours are primarily classified according to cell type into serous, mucinous, endometrioid, clear cell, transitional, and squamous cell tumours (1, 2, 8). However, none of these cells are found in the normal ovary and their development has long been attributed to Mullerian ‘neometaplasia of the ovarian surface epithelium (mesothelium). During embryonic life, the coelomic cavity is lined by mesothelium which also covers the gonadal ridge. The same mesothelial lining gives rise to Mullerian ducts, from which the fallopian tubes, uterus, and vagina arise. Thus, the tumour cells would resemble morphologically the epithelia of the fallopian tube, endometrium, or endocervix (1, 8). Recently, it has been hypothesized that cytokeratin 7-positive embryonic/stem cells would give rise to immunophenotypically distinct neoplastic progeny (9) which would support the old concept of ‘Mullerian neometaplasia. Besides the mesothelial origin, there is now compelling evidence that a number of what have been thought to be primary ovarian cancers actually originate in other pelvic organs and involve the ovary secondarily. It has been shown that some high-grade serous carcinomas arise from precursor epithelial lesions in the distal fimbriated end of the fallopian tube, whereas endometrioid and clear cell carcinomas originate from ovarian endometriosis (8).
Borderline tumours
Borderline tumours show epithelial proliferation greater than that seen in their benign counterparts and variable nuclear atypia; however, in contrast to carcinomas, there is absence of stromal invasion, and their prognosis is much better than that of carcinomas.
Serous borderline tumours generally occur in women aged 2050 years (average, 46 years). Serous tumours are more commonly bilateral (34%) than mucinous ones (6%) or other types. The tumours vary in size, although mucinous tumours may be gigantic.
Serous borderline tumours have one or more cysts lined to varying extents by papillary projections, ranging from fine and exuberant to grape-like clusters. These structures show (1) epithelial stratification, (2) moderate nuclear atypia, and (3) mitotic activity. By definition, the presence of more than focal microinvasion (i.e. discrete nests of epithelial cells <3 mm into the ovarian stroma) identifies a tumour as low-grade serous carcinoma (LGSC), rather than a borderline tumour (1, 2).Despite the lack of ovarian stromal invasion, serous borderline tumours, particularly those with exophytic growth, can implant on peritoneal surfaces and, rarely (about 10% of peritoneal implants), progress to LGSC and invade the underlying tissues. Histopathologically, invasive peritoneal implants and LGSC are identical lesions only distinguished by the timing of the disease and the volume of the tumour. Whereas invasive implants are early superficial lesions of microscopic or small macroscopic size (≤1- 2 cm), LGSC frequently presents as bulky disease (peritoneal carcinomatosis) (1, 2, 8).
Surgical cure is almost always possible if the serous borderline tumour is confined to the ovaries. Even if it has spread to the pelvis or abdomen, 90% of patients are alive after 5 years. Although there is a significant rate of late recurrence, the tumours rarely recur beyond 10 years. Late progression to LGSC has been reported in approximately 7% of cases (1, 2, 7). After fertility-sparing surgery, mucinous borderline tumours may ‘recur’ as carcinomas in the contralateral ovary; however, such tumours should be considered independent primary tumours (10, 11).
Malignant epithelial tumours (carcinomas)
Carcinomas ofthe ovary are most common in women aged 40-60 years, and are rare under the age of 35 years. Based on light microscopy and molecular genetics, ovarian carcinomas are classified into five main subtypes, which, in descending order of frequency, are high-grade serous carcinomas (>70%), endometrioid carcinomas (10%), clear cell carcinomas (10%), mucinous carcinomas (3-4%), and LGSCs (<5%) (8) (Table 70.2).
These subtypes, which account for 98% of ovarian carcinomas, can be reproducibly diagnosed and are inherently different diseases, as indicated by differences in epidemiological and genetic risk factors, precursor lesions, patterns of spread, molecular events during oncogenesis, responses to chemotherapy, and outcomes. With progress towards subtype-specific management of ovarian cancer, accurate subtype assignment is becoming increasingly important.Serous carcinomas
Molecular pathogenesis
Low-grade and high-grade serous carcinomas are fundamentally different tumours. Whereas low-grade tumours are frequently associated with serous borderline tumours and have mutations of KRAS or BRAF oncogenes, high-grade serous carcinomas lack ovarian precursor lesions and have a high frequency of mutations in TP53, but not in KRAS or BRAF. Interestingly, carcinomas arising in patients with germline BRCA1 or BRCA2 mutations (hereditary ovarian cancers) are almost invariably the high-grade serous type and commonly have TP53 mutations. An undetermined number of BRCAl- or BRCA2-related tumours arise from the epithelium of the
Pathology
High-grade serous carcinomas are the most common ovarian cancers and most patients present with advanced stage disease (approximately 80%). Two-thirds of serous cancers with extraovarian spread are bilateral. They are predominantly solid masses, usually with necrosis and haemorrhage and typically show obvious stromal invasion. Most tumours have a high nuclear grade with highly cellular papillae and solid areas (Figure 70.7a). The mitotic rate is very high. Psammoma bodies are often present (1, 2, 8).
LGSCs show irregular stromal invasion by small, tight nests of tumour cells within variable desmoplasia. The uniformity of the nuclei is the principal criterion for distinguishing low- and high-grade serous carcinomas (Figure 70.7b). LGSCs rarely progress to highgrade tumours (1, 2, 8).
Mucinous carcinoma
Molecular pathogenesis
Mucinous ovarian tumours are often heterogeneous.
Benign, borderline, non-invasive, and invasive carcinoma components may coexist within the same tumour. Such a morphological continuum suggests that tumour progression occurs from cystadenoma and borderline tumour to non-invasive, microinvasive, and invasive carcinomas. This hypothesis is supported by KRAS mutations in mucinous tumours: 56% of cystadenomas and 85% of carcinomas express mutated KRAS, with borderline tumours being intermediate (Table 70.2) (1, 8).Pathology
Mucinous carcinomas are usually large, unilateral, multilocular cystic masses containing mucinous fluid. They often exhibit papillary architecture (Figure 70.7c). Since benign and malignant components may coexist within a single specimen, these tumours should be sampled extensively. Mucinous tumours are bilateral in only 5%
Table 70.2 Main types of ovarian carcinoma
| I High-gradeserous | I Low-gradeserous | I Mucinous | I Endometrioid | I Clear cell | |
| Usual stage at diagnosis | Advanced | Early or advanced | Early | Early | Early |
| Presumed tissue of origin/ precursor lesion | Tubal metaplasia in inclusions of ovarian surface epithelium or fallopian tube | Serous borderline tumour | Adenoma-borderline- carcinoma sequence; teratoma | Endometriosis, adenofibroma | Endometriosis, adenofibroma |
| Genetic risk | BRCA1/2 | ? | ? | HNPCC | ? |
| Significant molecular abnormalities | p53 and pRb pathways | BRAF or KRAS | KRAS | PTEN β-catenin ARID1A PIK3CA | HNF1B ARID1A PIC3CA |
| K- RAS | |||||
| Microsatellite instability | |||||
| Proliferation | High | Low | Intermediate | Low | Low |
| Response to primary chemotherapy | 80% | 26-28% | 15% | ? | 15% |
| Prognosis | Poor | Favourable | Favourable | Favourable | Intermediate |
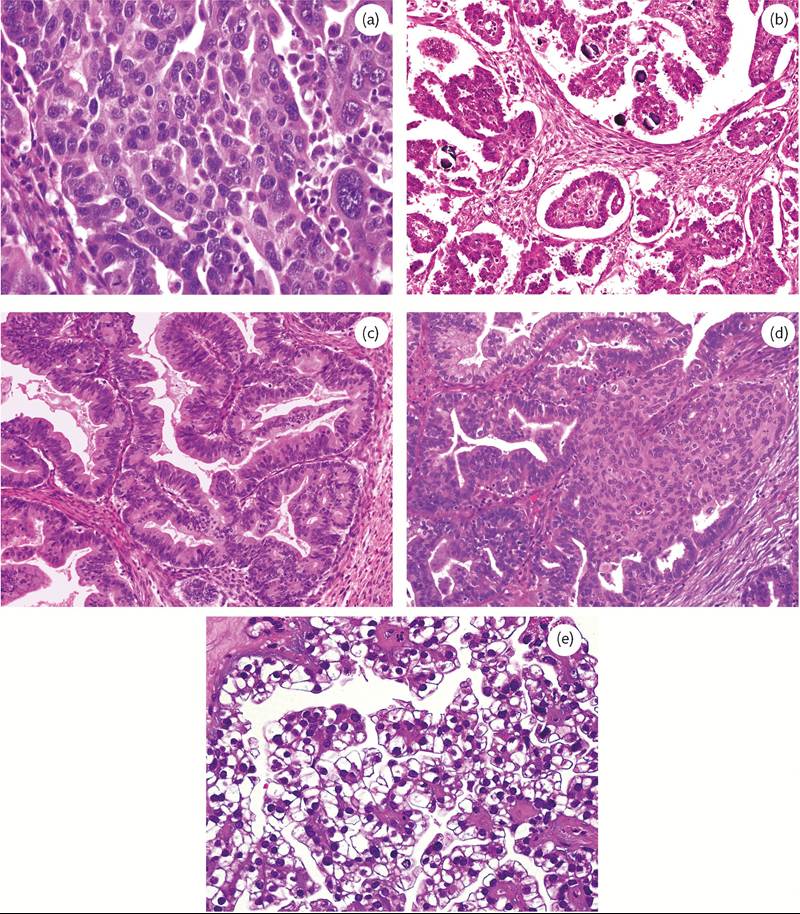
Figure 70.7 Representative examples of the five main types of ovarian carcinoma, which together account for 98% of cases: (a) high-grade serous carcinoma; (b) low-grade serous carcinoma; (c) mucinous carcinoma; (d) endometrioid carcinoma; and (e) clear cell carcinoma.
of the cases; thus, finding bilateral or unilateral mucinous tumours smaller than 10 cm should raise suspicion of metastases from a mucinous carcinoma elsewhere (e.g. gastrointestinal tract).
The category of mucinous borderline tumour with intraepithelial carcinoma is reserved for tumours that lack architectural features of invasive carcinoma but, focally, show unequivocally malignant cells lining glandular spaces. Mucinous borderline tumours with intraepithelial carcinoma have a very low likelihood of recurrence (1, 2, 8).
Mucinous carcinomas showing expansile or confluent glandular growth appear to have a more favourable prognosis than mucinous carcinomas with destructive stromal invasion. The combination of extensive infiltrative stromal invasion, high nuclear grade, and tumour rupture should be considered a strong predictor of recurrence for stage I mucinous carcinomas (1, 2, 8).
Pseudomyxoma peritonei is a clinical condition of abundant gelatinous or mucinous ascites in the peritoneum, fibrous adhesions, and frequently mucinous tumours involving the ovaries. The appendix is also involved by a similar mucinous tumour in 60% of the cases and appears normal in the remaining 40%. Current data suggest that in most cases the ovarian tumours are metastases from the appendiceal lesions (1, 2).
Endometrioid carcinoma
Endometrioid carcinoma histologically resembles its uterine counterpart (Figure 70.7d), may have areas of squamous differentiation, and is second only to serous carcinoma in frequency. It accounts for 10% of all ovarian cancers. These tumours occur most commonly after menopause. Up to half of these cancers are bilateral and, at diagnosis, most tumours are either confined to the ovary or within the pelvis (1, 2).
Molecular pathogenesis
Endometrioid carcinomas are thought to arise by malignant transformation of endometriosis, and not from ovarian surface epithelium. The most common genetic abnormalities in sporadic endometrioid carcinoma of the ovary are somatic mutations of the ARIDlA, beta-catenin (CTNNBl), and PTENgenes and microsatellite instability. Endometrioid borderline tumours also have CTNNBl mutations (Table 70.2) (8).
Pathology
Although they may be cystic, most endometrioid carcinomas are largely solid with areas of necrosis. These tumours are graded like their uterine counterparts. Between 15% and 20% of patients also harbour a uterine endometrioid carcinoma. Strong data suggest that most of these cases arise independently, although some may be metastases from one or the other. This distinction has important prognostic implications (1, 2).
Clear cell carcinoma
This enigmatic ovarian cancer is closely related to endometrioid adenocarcinoma, and often occurs in association with endometriosis. It constitutes 5-10% of all ovarian cancers usually occurring after menopause. The most common genetic abnormalities are somatic mutations of the ARIDlA, PTEN, and PIK3CA genes (Table 70.2) (1, 8).
Although patients typically present with stage I or II disease, clear cell carcinomas have a poor prognosis compared with other low-stage ovarian carcinomas. Clear cell carcinomas of the ovary resemble their counterparts in the vagina, cervix, and corpus; they show sheets or tubules of malignant cells with clear cytoplasm (Figure 70.7e).
Clinical features
By the time ovarian cancers are diagnosed, many have metastasized to (i.e. implanted on) the surfaces of the pelvis, abdominal organs, or bladder. Ovarian tumours have a tendency to implant in the peritoneal cavity on the diaphragm, paracolic gutters, and omentum. Lymphatic spread is preferentially to para-aortic lymph nodes near the origin of the renal arteries and to a lesser extent to external iliac (pelvic) or inguinal lymph nodes (1, 2).
Survival for patients with malignant ovarian tumours is generally poor. The most important prognostic index is the surgical stage of the tumour at the time it is detected (12). Overall, 5-year survival is only 35%. Prognostic indices for epithelial tumours also include histological type (grade) and the size of the residual neoplasm.
Surgery, which removes the primary tumour, establishes the diagnosis, and determines the extent of spread, is the mainstay of therapy. The peritoneal surfaces, omentum, liver, subdiaphragmatic recesses, and all abdominal regions must be visualized, and as much metastatic tumour removed as possible. Adjuvant chemotherapy is used to treat distant occult sites of tumour spread.
Germ cell tumours
Tumours derived from germ cells make up one-quarter of ovarian tumours. In adult women, ovarian germ cell tumours are virtually all benign (mature cystic teratoma or dermoid cyst), but in children and young adults, they are largely cancerous. In children, germ cell tumours are the most common ovarian cancer (60%); they are rare after menopause. Rarely, germ cell tumours may arise from preexisting somatic neoplasms of the female genital tract. In these cases, the teratoid tumours derive most likely from a pluripotent stem cell population of somatic neoplasms (1, 2).
Neoplastic germ cells may differentiate along several lines producing the following tumours:
• Dysgerminomas are composed of neoplastic germ cells, similar to oogonia of fetal ovaries
• Teratomas differentiate towards somatic (embryonic or adult) tissues.
• Yolk sac tumours form extraembryonic endoderm and mesenchyme and, less frequently, embryonic endodermal derivatives (intestine and liver).
• Choriocarcinomas feature cells similar to those covering the placental villi.
Malignant germ cell tumours in women older than 40 years usually result from transformation of one of the components of a benign cystic teratoma. Malignant germ cell tumours tend to be highly aggressive; however, with current chemotherapy, survival rates for many exceed 80% (1, 2).
Recent stem cell research has provided several highly diagnostic pluripotency markers, including transcription factors (SALL4, LIN28, OCT3/4, and SOX2) and cytoplasmic/membranous proteins (glypican-3) that are sequentially expressed in MGCTs according to their differentiation stage (1, 2).
Dysgerminoma
Dysgerminoma is the ovarian counterpart of testicular seminoma, and is composed of primordial germ cells. It accounts for less than 2% of ovarian cancers in all women. Most patients are between 10 and 30 years of age. The tumours are bilateral in about 15% of cases.
Pathology
Dysgerminomas are often large and firm and have a bosselated external surface. The cut surface is soft and fleshy. They contain large nests of monotonously uniform tumour cells that have clear glycogen-filled cytoplasm and irregularly flattened central nuclei. Fibrous septa containing lymphocytes traverse the tumour (1, 2).
Dysgerminomas are treated surgically; 5-year survival for patients with a stage I tumour approaches 100%. As the tumour is highly radiosensitive and also responsive to chemotherapy, even for higher- stage tumours 5-year survival rates still exceed 80%.
Teratoma
Teratoma is a tumour of germ cell origin that differentiates towards somatic structures. Most teratomas contain tissues from at least two, and usually all three, embryonic layers. Immature teratomas contain elements derived from the three germ layers. However, unlike mature cystic teratomas, immature teratomas contain embryonal tissues. These tumours account for 20% of malignant tumours in women under the age of 20. Microscopically, they show multiple components such as immature neural tissue (neuroepithelial rosettes and glia), glands, and other structures found in mature cystic teratomas. Grading is based on the amount of immature tissue present. Survival correlates with tumour grade (1, 2).
Yolk sac tumour
Yolk sac tumours are highly malignant neoplasms in women under the age of 30 years that histologically resemble the endoderm and mesenchyme of the primitive yolk sac (extra-embryonal) and embryonal somatic tissues (intestine and liver). They are typically large, with extensive necrosis and haemorrhage. The most common histotype is the reticular form. Schiller-Duval bodies are characteristic. They consist of papillae that protrude into spaces lined by tumour cells, resembling the glomerular spaces. The papillae are covered by a mantle of embryonal cells and contain a fibrovascular core and a central blood vessel.
Yolk sac tumours secrete alpha-fetoprotein. Detection of alphafetoprotein in the blood is useful for diagnosis and for monitoring the effectiveness of therapy. Once uniformly fatal, 5-year survival with chemotherapy for stage I yolk sac tumours exceeds 80% (1, 2).
Choriocarcinoma
Choriocarcinoma of the ovary is a rare tumour that mimics the epithelial covering of placental villi, namely, cytotrophoblast and syncytiotrophoblast. The pregnancy test is positive and the elevated serum level of human chorionic gonadotropin (hCG) may lead to precocious sexual development in young girls or menstrual abnormalities in older patients.
Sex cord/stromal tumours
These represent 10% of ovarian tumours, vary from benign to low- grade malignant, and may differentiate towards female (granulosa and theca cells) or male (Sertoli and Leydig cells) structures (1, 2).
Granulosa cell tumour
Granulosa cell tumours are the prototypical functional neoplasms of the ovary associated with oestrogen secretion. They should be considered low-grade malignancies because of their potential for local spread and the rare occurrence of distant metastases.
Most granulosa cell tumours occur after menopause (adult form) and are unusual before puberty. A juvenile form occurs in children and young women and has distinct clinical and pathological features (hyperoestrinism and precocious puberty).
Pathology
Adult-type granulosa cell tumours are large and focally cystic to solid. The cut surface shows yellow areas, due to lipid-rich luteinized granulosa cells, white zones of stroma, and focal haemorrhages. Random nuclear arrangement about a central degenerative space (Call-Exner bodies) gives a characteristic follicular pattern. Tumour cells secrete alpha- inhibin, a protein that suppresses pituitary release of follicle-stimulating hormone. Besides alpha-inhibin, calretinin and FOXL2 are the most important positive immunoreactions (1, 2).
Clinical features
Three-quarters of granulosa cell tumours secrete oestrogens. Thus, endometrial hyperplasia is a common presenting sign. Endometrial adenocarcinoma may develop if a functioning granulosa cell tumour remains undetected. At diagnosis, 90% of granulosa cell tumours are within the ovary (stage I). Over 90% of these patients survive 10 years. Tumours that have extended into the pelvis and lower abdomen have a poorer prognosis. Late recurrence after surgical removal is not uncommon after 5-10 years and is usually fatal (1, 2).
Sertoli-Leydigcell tumours
Ovarian Sertoli-Leydig cell tumours are rare androgen-secreting mesenchymal neoplasms of low malignant potential that resemble embryonic testis. Tumour cells typically secrete weak androgens (dehydroepiandrosterone). Sertoli-Leydig cell tumours occur at all ages but are most common in young women of childbearing age. They vary from well to poorly differentiated and some have heterologous elements (e.g. mucinous glands and, rarely, even skeletal muscle and cartilage).
Nearly half of all patients with Sertoli-Leydig cell tumours exhibit signs of virilization. Initial signs are often defeminization, manifested as breast atrophy, amenorrhea, and loss of hip fat. Once the tumour is removed, these signs disappear or at least lessen. Well- differentiated tumours are virtually always cured by surgical resection, but poorly differentiated ones may metastasize (1, 2).
Steroid cell tumour
Steroid cell tumours of the ovary, also called lipid cell tumours, are composed of cells that resemble lutein cells, Leydig cells, and adrenal cortical cells. Most steroid cell tumours are hormonally active, usually with androgenic manifestations.
Tumours metastatic to the ovary
About 3% of cancers found in the ovaries arise elsewhere, mostly in the large intestine, breast, endometrium, and stomach, in descending order. These tumours vary from microscopic lesions to large masses. Metastatic tumours large enough to cause symptoms originate most often in the colon.
Krukenberg tumours are metastases to the ovary, composed of nests of mucin-filled ‘signet-ring’ cells in a cellular stroma derived from the ovary. The stomach is the primary site in 75% of cases and most of the rest are from the colon (1, 2).
Bilateral ovarian involvement and multinodularity suggest a metastatic carcinoma, and both ovaries are grossly involved in 75% of cases.