ACYANOTIC CHD
•
• In large shunts, increased inflow on right side via Lt gt; Rt shunts may lead to right sided volume overload and increased pulmonary blood flow, which manifests with ill-sustained sucking, recurrent chest infections or frank CCF.
• Persistent elevation of pulmonary blood flow may produce secondary changes in pulmonary vasculature, leading to development of pulmonary arterial hypertension (PAH). Over the years, rising pulmonary pressure may equalize and then exceed the systemic pressure, leading to reversal of shunt-flow, i.e. Eisenmengerization with appearance of cyanosis.
Acyanotic CHDs with pressure overload are obstructive lesions, characterized by ventricular outflow obstruction (AS, PS, CoA) and clinical presentation depends on magnitude of obstruction.
Some common acyanotic heart diseases have been discussed in this section.
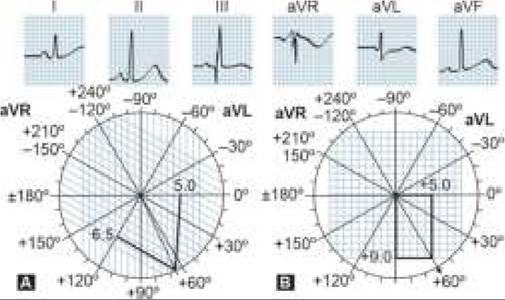
Fig. 17.14: Hemodynamics: VSD.
(1) VSD; (2) RV Volume overload; (3) PAH; (4) #8593;PBF; (5) #8593;Flow across mitral valve, (6) LVH. Double arrows indicate extra blood flow.
1. Ventricular Septal Defect (VSD)
VSD is the commonest cardiac malformation, accounting for ~25% of CHDs. It is also an integral part of many cyanotic CHDs, though present section deals with only isolated defects.
Anatomically, 90% of VSDs are located in the membranous portion of interventricular septum (membranous VSDs), while rests are in muscular part (muscular VSDs). Size-wise, VSDs may be classified as small or restrictive (lt;0.5 cm2), moderate (0.5-1.0 cm2) or large (gt;1.0 cm2). VSDs with inter-ventricular pressure gradient of gt;64 mm Hg are also termed restrictive VSDs.
Hemodynamics and consequent clinical presentation in VSD depend on its size and magnitude of shunt across the ventricles, as discussed below and graphically presented in Fig.
17.14.• Primary hemodynamic defect in VSD is a transseptal LV gt; RV shunt due to higher pressure in LV (100 / 70 mm Hg) than in RV (30/10 mm Hg). Since this pressure gradient persists throughout the systole, typical murmur is pansystolic (PSM) in left parasternal area (3-4th space), which is louder in smaller VSDs (due to higher flow turbulence across a smaller gap). At birth, shunt is minimal due to higher pulmonary resistance/RV pressure and murmur is absent, which usually appears by the end of first week due to gradual drop in pulmonary resistance after expansion of lungs and consequent increase in shunt flow.
• Increased RV volume due to trans-VSD flow, leads to—(a) CCF after variable time-gap, (b) ESM in pulmonary area due to increased pulmonary blood flow, and (c) loud/palpable P2 due to secondary PAH. However, RVH is uncommon despite PAH, as systolic shunt-flow in VSD is mainly directed towards the pulmonary valve, when RV is also contracting.
• Increased pulmonary blood flow leads to pulmonary congestion with feeding difficulties, recurrent chest infections and pulmonary plethora on chest skiagram.
• Increased pulmonary blood flow also enhances venous return to LA and flow across the mitral valve, producing a late or mid diastolic murmur, due to increased flow across a normal-size valve (functional MS).
• Increased in-flow in LV from LA as well as the need for stronger contractions to maintain adequate cardiac output in view of LVgt;RV leak, leads to LV volume overload and LVH respectively with hyperdynamic apex-beat, shifted downwards and outwards.
• Early closure of aortic valve due to LV leak through VSD and late closure of pulmonary valve due to additional blood volume in RV via shunt, leads to wide splitting of second HS.
In small VSDs, pathology is usually limited to a small shunt (loud murmur) without significant hemodynamic changes.
Clinical manifestations depend on the size of the defect. While small VSDs may remain asymptomatic in childhood and diagnosed accidentally, large VSDs may present even in neonatal period.
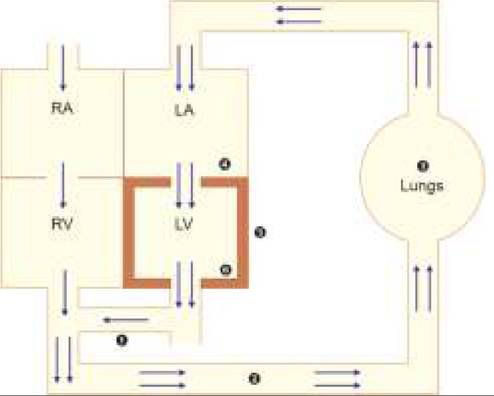
Most cases present by 6-10 weeks, with symptoms suggestive of increased pulmonary blood flow, e.g. feeding difficulties (suck-rest- suck cycle) and recurrent chest infections. Important clinical signs are given in Table 17.17.Size of the VSD may be clinically assessed by—(a) age of presentation, (b) magnitude of symptoms, (c) loudness of PSM, (d) loudness of P2, and (e) severity of PAH. Asymptomatic/late-manifesting VSD with loud PSM but without loud P2 or significant PAH indicates small VSD. Diagnosis depends on:
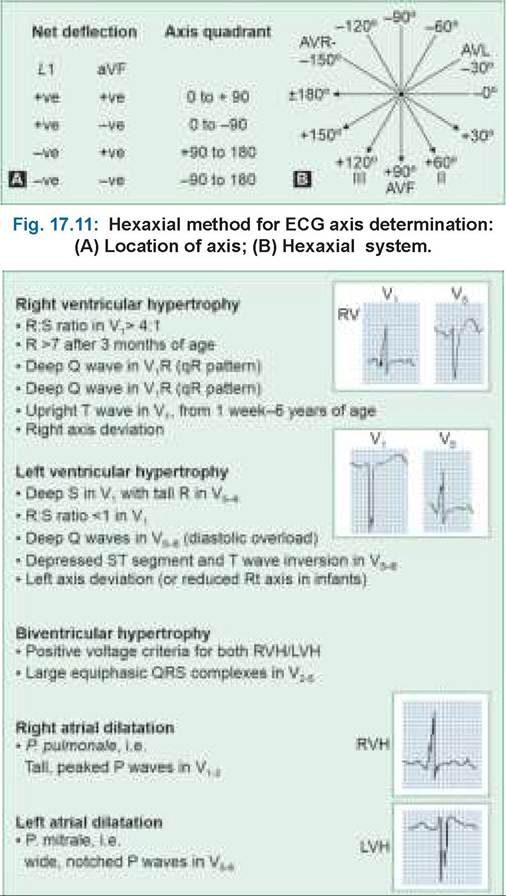
• Chest X-ray suggestive of LVH with pulmonary plethora.
*characteristic clinical sign
• ECG suggestive of LVH or biventricular hypertrophy.
• Echocardiography with Doppler studies is essential to—(a) visualize position/size of VSD, (b) estimate shunt-size by assessing the volume overload in LV/ LA and (c) assess severity of PAH.
• Cardiac catheterization is required before surgery to identify exact hemodynamic status.
Course and complications: Natural course depends on the size of VSD and includes:
• Spontaneous closure by 2-3 years in 30-50% cases, though remaining VSDs may also reduce in size. Small, muscular VSDs are more likely to close than large and membranous VSDs. Spontaneous closure after 3 years of age is unlikely (lt;10%).
• Recurrent CCF, usually by 2-3 months in large VSDs and by late infancy in moderate defects.
• Infective endocarditis is more common in small VSDs, due to small but more forceful shunt flow causing jet injury on RV walls.
• Development of subvalvular PS, due to hypertrophy of RV infundibulum in ~10% cases, which may increase RV pressure with reduce shunt flow and symptomatic improvement.
• Development of pulmonary vascular obstructive disease following long-standing hyperdynamic pulmonary hypertension in moderate/large defects.
• Aortic valve prolapse with aortic regurgitation, specially in supracristal VSDs due to loss of support to the coronary cusp.
• Eisenmengerization, i.e. reversal of shunt in large VSDs or those with associated PS, due to gradual rise in RV pressure and reversal of pressure-gradient across the ventricles. Clinically, these cases may show temporary symptomatic improvement with disappearance of murmur due to reduction in Rtgt;Lt shunt before appearance of cyanosis due to reversed Ltgt;Rt shunt.
Management involves closure of the defect by open surgery (Patch closure) or interventional device closure, as follows:
• Large VSDs with refractory CCF or severe PH must be operated as early as possible, while those with controlled CCF must be operated by 6 months.
• Moderate size VSDs should be operated by 1-2 years, if controlled by medications. However in asymptomatic cases, surgery is generally deferred till 2-5 years of age in expectation of the spontaneous closure.
• Surgery is not indicated in small VSDs (lt;3 mm) unless they develop aortic valve prolapse, progressive RV outflow obstruction or Infective endocarditis.
• All VSDs, irrespective of the size or severity, should be operated as soon as possible in case of aortic cusp prolapse or significant aortic regurgitation.
• Surgery is contraindicated in cases with severe PH or irreversible pulmonary vascular disease and may be difficult in multiple or inaccessible VSDs, who may benefit by pulmonary artery bending procedures.
• Device closure is preferred in cases weighing gt;8 kg with adequate rim around the defect including—(a) mid-and anterior-muscular VSDs, (b) post-operative residual VSD, and (c) perimembranous VSDs at least 4 mm away from aortic valve. Device closure is contraindicated in cases with associated AR, conduction abnormalities, who should be treated surgically.
• After device closure, patient has to be on follow-up with anti-platelet therapy and infective endocarditis prophylaxis for 6 months. IE prophylaxis is also indicated after surgery.
• Operative mortality is lt;2% and complications include heart block and re-opening of defect.
Medical management till surgery includes: (a) regular follow-up, (b) early management of CCF, infections, anemia, etc., and (c) infective endocarditis prophylaxis.
2. Patent Ductus Arteriosus (PDA)
Patent ductus arteriosus (PDA) denotes persistence of fetal communication between left pulmonary artery and descending aorta, distal to the origin of left subclavian artery. Normally, this communication (ductus arteriosus) closes functionally within 10-12 hours of birth and anatomically by 4-6 weeks (Ligmanetum arteriosum).
Isolated PDA accounts for ~6-8% of CHDs. In addition, PDA is also present as life-saving component in many obstructive and cyanotic CHDs, offering alternative route for pulmonary blood flow.
PDA is three times more common in females than in males. It is also frequently present in extreme preterms and closes spontaneously with maturity, indicating merely a developmental delay. In term newborns, it is a structural defect in the muscular wall, unlikely to close spontaneously or by medical therapy.
Hemodynamics and consequent presentation in PDA depends on the magnitude of shunt, as discussed below and graphically presented in Fig. 17.15.
• Primary hemodynamic defect in PDA is trans-ductal Lt gt; Rt shunt, due to higher pressures in descending aorta (120/80 mm Hg) than in left PA (25/05 mm Hg). As this pressure gradient persists during systole as well as diastole, PDA murmur is typically a continuous machinery murmur in left infraclavicular region, more marked in systole.
• The shunt is minimal at birth due to high PA pressure and PDA murmur generally appears after 24-48 hours, corresponding to gradual expansion of lungs and fall in PA pressure. In presence of significant PAH, murmur may only be systolic due to equalization of aortic and pulmonary pressure in diastole.
• Increased pulmonary blood flow due to extra-flow received via ductus, leads to pulmonary congestion with recurrent respiratory infections and feeding difficulties; and subsequently development of PAH with loud P2 and ESM at pulmonary area.
• Increased venous return from congested lungs leads to LA volume overload and dilatation.
• Increased LA volume increases flow across the mitral valve to produce loud S1, delayed diastolic murmur at apex and occasionally, the S3. Intensity of S1 and this murmur correlates directly with severity of shunt.
• Increased flow from LA as well as the need for stronger LV contractions to maintain adequate systemic circulation (in view of leak form aortic side at ductus) leads to LV volume overload and LVH respectively, p resenting with hyperdynamic apex beat, shifted downwards and outwards.
• LV volume overload increases LV emptying time, leading to delayed A2 with narrow, absent or reverse (paradoxical) split of S2. Increased LV outflow though a normal size aortic valve may also produce an ESM at aortic area as well as an ejection click due to dilatation of aortic root. However, this ESM is generally drowned in continuous murmur.
• Leak from aortic side via ductus leads to rapid drop in aortic pressure during ventricular diastolic phase, with water-hammer pulse and wide pulse pressure.
Clinical manifestations depend on the magnitude of shunt and very small PDA may remain asymptomatic or manifest in late childhood. Most cases present at 6-10 weeks with symptoms suggestive of increased pulmonary blood flow, e.g. feeding difficulties (suckrest-suck cycle), recurrent chest infections, failure to thrive or frank CCF. Important clinical signs are given in Table 17.18.
Diagnosis depends on:
• Chest X-ray suggestive of LVH with pulmonary plethora. Aortic knuckle may be prominent due to dilatation of the aortic root.
• ECG suggestive of LVH with/without strain pattern (deep Q and tall T waves in left chest leads).
• Echocardiography helps to visualize the shunt and assess ventricular sizes and pressures.
Course and complications: Spontaneous closure is possible in preterms and may be hastened by indomethacin therapy. Organic PDA does not close spontaneously or by medical therapy and can lead to:
• CCF at any age after first 24-48 hours,
• Infective endocarditis with vegetations on pulmonary side of shunt
• Rarely, Eisenmengerization due to severe PAH. In Eisenmengerization of PDA, cyanosis is typically differential-more prominent in lower limbs due to ductal mixing beyond the left subclavian artery.
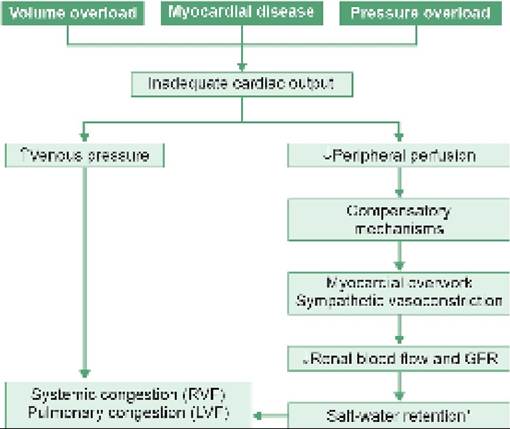
Fig. 17.15: Hemodynamics: Patent ductus arteriosus.
1. PDA; 2. PAH; 3. #8593;PBF, 4. #8593;Flow across mitral valve, 5. LVH, 6. #8593;LV outflow. Double arrows indicate extra blood flow.
Management may be divided into:
• Supportive therapy with adequate nutrition, early diagnosis/treatment of CCF and chest infections, regular follow-up and prevention of infective endocarditis.
• Medical closure is possible only in preterms, by IV/PO Indomethacin, a prostaglandin synthetase inhibitor (0.1 mg/kg/dose), given as three doses at 12-hourly intervals. It is contraindicated in babies with hepatic/renal disease, bleeding tendencies or ductus-dependent cyanotic lesions. Ibuprofen (PO, three doses as 10 mg/kg gt;5 mg/kg gt;5 mg/kg at 12-hr intervals) may also be used.
• Surgical closure is indicated in all cases including failed medical closure in preterms, either by open surgical ligation or preferably by transcatheter devices as follows:
± Closure is recommended by 3 months in large PDAs or moderate PDAs with CCF, 6-12 months in moderate PDAs without CCF and 12-18 months in smaller defects.
± Closure intervention is contraindicated in cases with irreversible pulmonary vascular disease.
± Device closure is preferred in babies weighing gt;6 kg while surgical ligation is advisable in lighter cases or those with unusual PDA shape, progressive/ symptomatic aneurysm or endarteritis. Operative mortality is lt;1%.
± Infective endocarditis prophylaxis is indicated during next 6 months.
3. Atrial Septal Defects (ASD)
Isolated ASDs account for ~8-10% of CHDs, twice more common in females. In addition, ASD is frequently present in various cyanotic CHDs. Exact incidence may be much higher, as many ASDs remain undetected throughout the life.
Patent foramen ovale (PFO) is a common echocardiographic finding, usually of no hemodynamic significance and is not considered as ASD. However, PFO may play an important role if other structural heart defects are present. It also provides a route for paradoxical systemic embolization.
Anatomically ASD may be divided into:
• Common Ostium secundum (OS) defects (70-80%), at the site of fossa ovalis. In practice, the term ASD is used only to denote these defects, discussed here.
• Uncommon Ostium primum defects (15-20%) or endocardial cushion defects in lower atrial septum below the fossa ovalis, frequently associated with clefts in mitral or tricuspid valve leaflets.
• Rare sinus venosus defects (5-10%) located in postero- superior septum at the site of superior vena caval entry into RA.
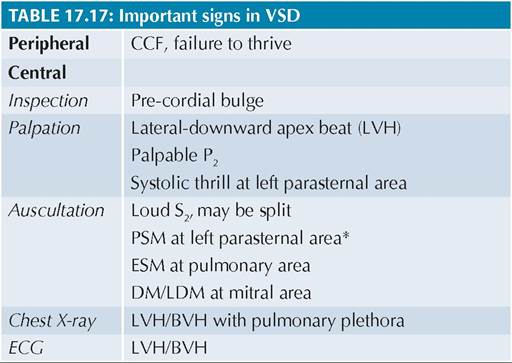
Fig. 17.16: Hemodynamics: Atrial septal defect
1. ASD; 2. RA Volume overload; 3. RVH; 4. PAH; 5. #8593;PBF
Note: 3, 4. and 5. develop after many years. Double arrows indicate extra blood flow.
Hemodynamics in common OS-ASD depend on the size of defect and relates more to the volume changes in atria rather than pressure changes, due to relatively compliant and less muscular atrial chambers. Most cases do not manifest till second decade, unless having very large defects. Important hemodynamic changes are discussed here as well as graphically presented in Fig. 17.16.
• Primary hemodynamic defect in OS-ASD is Lt gt; Rt shunt at the atrial level, which is usually minimal and silent due to nearly comparable pressures in atria (0-5 mm).
• In large defects, significant shunt leads to RA volume overload and dilatation (tall peaked P waves in V1_2 on ECG) as well as increased flow across the tricuspid valve, producing late diastolic rumbling murmur at tricuspid area.
• Increased flow into RV produces volume overload and some RVH on X-ray/ECG, but less obvious clinically.
• RV volume overload increases flow across the pulmonary valve, producing ESM at pulmonary area, delayed P2 and consequently splitting of second HS. As RV is already overloaded, further increase in RV volume during inspiration, as seen normally, is not possible, leading to fixed split of S2.
• After many years, usually in second decade, RV volume overload may lead to CCF and/or increased pulmonary blood flow, with onset of clinical manifestations.
Clinical manifestations: Isolated ASD is usually asymptomatic in childhood, diagnosed accidentally. Most cases manifest in late adolescence or early adulthood with features of increased pulmonary blood flow, e.g. effort intolerance or recurrent chest infections. Important clinical signs are given in Table 17.19.
TABLE 17.19: Important signs in ASD
Diagnosis is supported by:
• Chest X-ray suggestive of RVH with normal pulmonary blood flow. Sometimes, pulmonary conus may be prominent due to pulmonary artery dilatation. Pulmonary blood flow may be minimally increased in large defects or older children.
• ECG showing RVH with right-axis deviation and rsR' pattern (pathognomonic in ~90%) in right chest leads suggestive of right bundle branch block.
• Echocardiography reveals—(a) exact location and size of the defect in subcostal 4-chamber view, (b) abnormalities in RA, RV and PA size, (c) paradoxical motion of intra-atrial septum on M-mode. Doppler studies reveal characteristic flow pattern with maximum shunt during diastole.
Course and complications: Nearly all ASDs lt;3 mm close spontaneously by 2 years, while those gt;8 mm are unlikely to close. Complications, e.g. CCF or PAH are rare before third decade, except arrhythmias, e.g. sick sinus syndrome. Infective endocarditis is very rare in isolated ASD and prophylaxis is not required.
Management involves surgical or device closure, as follows:
• All asymptomatic ASDs should be operated at 2-4 years of age, except that the surgery in sinus venosus defects may be delayed till 4-5 year. ASDs diagnosed later than recommended age for surgery should be operated soon after the diagnosis.
• Earlier closure is recommended in symptomatic ASDs with CCF and PH, after ruling out associated cardiac defects.
• Surgery is contraindicated in rare cases with severe PH or irreversible pulmonary vascular disease.
• Device closure is preferred for OS-ASD with good rim #8805; 5 mm, while others may need surgical repair with operative mortality of lt; 1%.
• After device closure, patient has to be on follow-up with anti-platelet therapy and infective endocarditis prophylaxis for 6 months. IE prophylaxis is also indicated after surgery.
4. Aortic Stenosis (AS)
Aortic stenosis (AS) may be congenital or rheumatic, though later is extremely rare in childhood. Congenital AS accounts for 5-8% of CHDs, more common in males. Structurally, congenital AS may be valvular, supravalvular or subvalvular in location.
• Valvular AS due to bicuspid aortic valve is the commonest CHD in adults, present in ~2% population.
• Subvalvular stenosis is usually seen with other CHDs or hypertrophic cardiomyopathy.
• Supravalvular stenosis is seen in William syndrome — with Elfin-facies, mental retardation and idiopathic hypocalcemia.
Clinical presentation depends on the site and severity of LV outflow obstruction. While mild cases are usually asymptomatic, severe AS present with easy fatigability and syncopal attacks, due to lower cardiac output. Important signs include:
• Low-volume pulse and narrow pulse pressure due to reduced LV output,
• A diamond-shaped, ejection systolic murmur in right second intercostal space due to LV outlet obstruction, which is typically referred to the neck,
• An ejection click just before the ESM, due to poststenotic dilatation of ascending aorta in valvular lesions.
• Heaving apex beat, displaced downward and laterally due to systolic LV overload and LVH
• Narrow or reverse splitting of S2, due to increased LV ejection time gt; delayed AV closure.
Diagnosis is supported by—(a) chest skiagram showing LVH with dilatation of aortic knuckle, (b) ECG suggestive of LVH, and confirmed on (c) echocardiography.
Management: Asymptomatic mild AS need no intervention except avoiding strenuous exercise, which may precipitate hemodynamic decompensation.
Symptomatic cases need either—(a) balloon aortic valvuloplasty for valvular/subvalvular lesions, (b) surgical valvotomy or (c) valve replacement, as follows:
• Valvular or supravalvular AS needs immediate surgery/balloon dilatation in newborns with critical AS or severe LV dysfunction, while elective balloon dilatation is indicated in cases with peak gradient gt;64 mm Hg, mean gradient gt;40 mm Hg, symptomatic AS or ST changes on ECG even with lower gradients.
• Subvalvular AS needs surgery if (a) peak gradient is 50 mm Hg or (b) peak gradient is lt;50 mm Hg but with associated moderate to severe AR or LV dysfunction or (c) plans to involve in competitive sports
• Infective endocarditis prophylaxis is indicated for next 6 months after valvuloplasty or life-long in cases of prosthetic valve replacement.
4. Pulmonary Stenosis (PS)
Pulmonary stenosis (PS) may be an isolated defect or a component of other CHDs. It is the commonest CHD in Noonan syndrome.
Structurally, It may be valvular (commonest), supravalvular or infundibular (subvalvular). Co-existing hypoplasia/stenosis of distal pulmonary vessels is common and needs to be excluded before surgery.
Clinical presentation depends on the severity of RV outflow obstruction. While mild cases are usually asymptomatic, moderate to severe PS presents with CCF at variable age. Important signs include:
• A diamond shaped ESM in pulmonary area, due to outflow obstruction,
• Soft or absent P2 in valvular PS, though it may be louder in supravalvular PS.
• Wide splitting of S2 due to-prolonged RV ejection time gt; delayed PV closure.
• An ejection click in pulmonary area indicates valvular PS with post-stenotic dilatation of PA.
• Lateral shifting of apex beat and left parasternal heave, due to RVH.
Diagnosis is supported by—(a) chest X-ray showing RVH, oligemic lung fields and absence or post-stenotic dilatation of pulmonary conus, (b) ECG suggestive of RVH, and (c) chocardiography to assess the severity.
Treatment of choice is the Balloon dilatation with/without stenting, alternative being surgery with/without valve replacement, as follows:
• All cases needs immediate intervention in newborns with critical PS or severe LV dysfunction, while elective balloon dilatation is indicated in cases with— (a) peak gradient gt;64 mm Hg, (b) mild RV hypoplasia causing hypoxia or (c) dysplastic pulmonary valve.
• All cases of peripheral PA or branch stenosis with—(a) gt;50% diameter narrowing, (b) RV systolic pressure gt;50 mm Hg, or (c) gt;20% difference in perfusion of both lungs on perfusion scan also need balloon dilatation with/without stenting or surgery.
• Infective endocarditis prophylaxis is indicated for next 6 months after valvuloplasty or life-long in cases of prosthetic valve replacement.
5. Coarctation of Aorta (COA)
Coarctation of aorta (CoA), i.e. localised or diffuse narrowing of aorta from any point between transverse arch to iliac bifurcation, contributes to ~2-3% of CHDs in children. CoA is twice more common in males, sometimes associated with Turner or Noonan syndrome.
Over 70% CoA also have VSD or left-sided lesions, e.g. bicuspid aortic valve, mitral valve abnormalities, etc., together referred as Shone complex.
Anatomically, all CoA may be broadly divided into two types, with significant differences in hemodynamics and clinical presentations:
a. Juxtaductal CoA accounts for ~98% CoA, characterized by coarctation at the level of ductus arteriosus, just below the origin of left subclavian artery.
b. Pre-ductal or infantile CoA (lt;2%), characterized by localized coarctation or diffuse tubular hypoplasia before the ductus arteriosus.
Hemodynamically two facts are worth considerations:
a. Upper half of the body is supplied by preductal branches of ascending/transverse aorta (innominate, left carotid/subclavian), while lower body is supplied by post-ductal branches of descending aorta. In CoA, deficient flow in descending aorta leads to development of:
- Pulse-BP deficit between upper and lower body,
- Renal ischemia and consequent hypertension, and
- Collaterals between preductal and postductal vessels to augment distal blood flow.
b. In utero, upper body receives more oxygenated preductal flow from LV, while distal body is largely supplied by less oxygenated blood through RV gt; PDA gt; descending aorta.
- In juxtaductal CoA, coarcted segment interferes with ductal flow, leading to development of collaterals in fetal life itself, which partially sustain the distal flow after postnatal ductal closure. Hence, most cases manifest in later life with hypertension (Fig. 17.17).
- In preductal CoA, coarcted segment does not obstruct the ductal flow and hence, collaterals do not develop in fetal life. Absence of collaterals at the time of birth and postnatal ductal closure in these cases leads to—(a) sudden drop in descending aortic pressure, with hypertension, and (b) sudden increase in proximal aortic pressure with LVH.
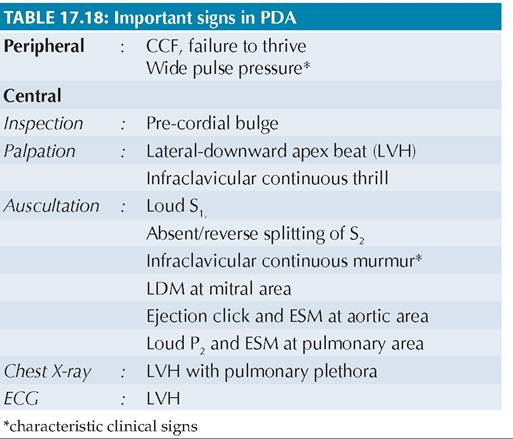
Fig. 17.17: Juxtaductal coarctation of aorta.
Consequently, most cases or preductal CoA present with severe hypertension, LVH and CCF in neonatal period or early infancy. In some cases, low aortic pressure in post-coarcted segment or severe pulmonary arterial hypertension may facilitate longer patency of PDA with Rt gt; Lt shunt, leading to differential cyanosis in lower limbs.
Hemodynamic changes in CoA are further altered by presence of co-existing heart lesions, e.g. VSD.
Clinical spectrum of CoA varies according to the severity and location of defect, as well as presence of coexisting cardiac lesions.
Uncomplicated Juxtaductal CoA, usually presents with asymptomatic or symptomatic hypertension in late childhood, though some cases may complain of weakness and leg pains after exercise (? due to ischemia). Important clinical signs include:
• Systolic hypertension, more prominent on exercise.
• Pulse/BP disparity between upper and lower limbs, more prominent on exercise and presents with:
- Absent/poor pulsations in lower limb vessels, e.g. femoral, popliteal or dorsalis pedis artery;
- Radiofemoral delay, due to slower filling of descending aorta by collaterals rather than by direct aortic flow,
- Less BP in lower limbs than upper limbs (normally, lower limb BP exceeds upper limb BP by 10-20 mm Hg). Lower BP in left arm than in right arm suggests involvement of left subclavian artery in coarcted segment.
• Long systolic or continuous murmurs over chest and back due to collaterals.
• Cardiac signs depend on severity of obstruction and co-existing heart lesions, ranging from normal precordium to:
- Signs of aortic flow obstruction, e.g. prominent arterial pulsations in neck and/or ejection systolic murmur in interscapular area.
- Signs of LV overload, e.g. loud A2, ejection click or LVH (down/outward heaving apex beat).
- Signs of other heart lesions, e.g. aortic ESM/ LDM (bicuspid aortic valve), mitral MDM (MS), etc.
Preductal CoA presents in neonatal period with cyanosis/differential cyanosis and CCF, though some cases have delayed presentation after the closure of ductus. Pulse/BP disparity is less prominent in these cases and collateral murmurs are rare.
Complications: CCF is the most important complication of CoA in infancy, in cases with preductal/severe juxtaductal disease or co-existing heart lesions. Untreated cases of moderate/mild CoA are usually asymptomatic in childhood except potential risk of infective endocarditis with vegetations on aortic wall or bicuspid aortic valve.
Most cases develop hypertensive complications in 2nd- 3rd decade, e.g. (a) CCf, (b) hypertensive encephalopathy, (c) premature coronary disease, (d) aortic aneurysms. Cerebrovascular accidents are common in cases with coexisting intracranial aneurysms, e.g. Berry's aneurysm. Diagnosis is clinically indicated by hypertension with pulse-BP deficit and supported by:
• Chest X-ray may be normal in early life. Late signs include—(i) mild LV cardiomegaly, (ii) prominent aortic knuckle, (iii) pre-/post-stenotic dilatation of aorta, and (iv) notching of the lower rib borders due to collaterals.
• Barium swallow may reveal characteristic E sign, due to pre-and post-stenotic dilatation.
• ECG may be normal or reveals LVH.
• Echocardiography, for—(i) direct visualization of coarctation through suprasternal approach, (ii) detection of co-existing heart lesions.
• Color Doppler studies to assess the—(i) severity of coarctation by recording pre-stenotic vs post-stenotic pressure gradient, (ii) status of collaterals.
• Cardiac catheterization to detect additional heart defects and adequacy of collateral flow, before surgery in established case.
Management of CoA is essentially surgical, after stabilization with supportive medical treatment, e.g. treatment of CCF. Infants with pre-ductal or severe juxtaductal CoA are usually ductus-dependent and may require prostaglandin E1 infusions to re-open or keep the ductus patent till surgery is arranged. Important principles of surgical management include:
• All cases of CoA with peak gradient 20 mm Hg, LV dysfunction or LVH, hypertension or gt;50% narrowing relative to aortic diameter at diaphragm on imaging studies need intervention
• Cases with significant LV dysfunction, CCF or severe hypertension should be operated immediately, while others must be operated as soon as possible, i.e. between 3 months to 2 years. Surgical risk increases with advancing age due to deteriorating LV function and degenerative changes in aortic wall.
• Balloon angioplasty is generally preferred over surgical aortoplasty due to less operative morbidity, mortality and costs, though carries higher risk of aneurysm formation. Surgical aortoplasty is necessary in newborns with aortic arch hypoplasia.
Children gt; 25 kg or those with post-operative recoarctation may need stenting, which is contraindicated lt; 10 years of age.
• Important post-operative complications include—(a) transient rebound hypertension in immediate postoperative period, and (b) late re-coarctation, specially in cases operated in infancy. Infective endocarditis prophylaxis is indicated for next 6 months.
17.5.3