CYANOTIC CHDs
Cyanotic CHDs (Rt gt; Lt shunts) may be broadly divided into two categories—(a) those with decreased pulmonary blood flow (PBF), and (b) those with increased PBF.
Cyanotic CHDs with decreased PBF essentially have two lesions—an obstruction to PBF at tricuspid or pulmonary valve level (usually PS), and a Rt gt; Lt shunt at atrial or ventricular level.
Common lesions of this group include tetralogy of Fallot (TOF), tricuspid atresia, pulmonary atresia, and total anomalous venous drainage (TAPVR) with PS.Severity of cyanosis in these cases depends on the degree of PBF obstruction and status of ductus arteriosus that provides alternate route to PBF. These newborns are usually not-cyanotic during first few days of life due to ductal patency and cyanosis appears as the ductus closes.
Cyanotic CHDs with increased PBF are characterized by Rt gt; Lt shunt but without PBF obstruction (no PS). In these cases, severity of cyanosis depends on the size of shunt and most cases present with a combination of cyanosis and CCF. Common lesions in this group include transposition of great arteries (TGA), truncus arteriosus, single ventricle, or TAPVR without PS.
Apart from cyanosis and CCF, other common complications of cyanotic CHDs include:
• Compensatory polycythemia, leading to thromboembolic complications and Iron deficiency anemia,
• Metabolic complications due to hypoxic tissue injury and increased cell mass, e.g. (i) hyperuricemia and gout, (ii) hyperkalemia, (iii) hyperphosphatemia, (iv) hypocalcaemia (secondary to hyperphosphatemia), etc.
Some common cyanotic CHDs are as follows:
1. Tetralogy of Fallot (TOF)
Tetralogy of Fallot (TOF) is the commonest cyanotic CHD, accounting for gt;8-10% of all CHDs.
Anatomically it is characterized by four components- two primary lesions: (a) pulmonary stenosis, usually of infundibular type, and (b) ventricular septal defect, usually peri-membranous; as well as two secondary lesions, (c) overriding of the aorta, receiving blood from both ventricles, and (d) right ventricular hypertrophy.
Associated defects include hypoplasia of pulmonary or coronary vessels, PDA or right-sided aortic arch.
Fallot physiology is a term used to describe certain cyanotic CHDs with decreased PBF and ToF-like clinical presentation, ECG and X-ray abnormalities, e.g. (a) TGA with VSD and PS, (b) double outlet right ventricle (DORV) with VSD and PS, (c) single ventricle with PS, (d) tricuspid atresia with diminished PBF. Echocardiography is essential to distinguish between these disorders.
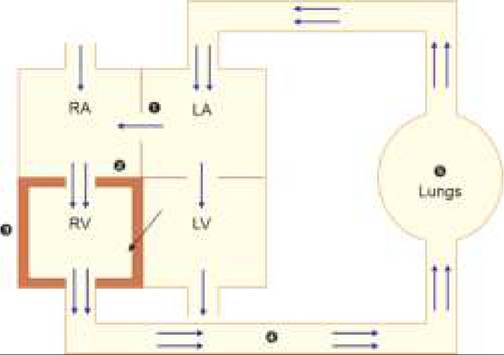
Fig. 17.18: Hemodynamics: Tetralogy of Fallot
1. VSD; 2. Overriding aorta; 3. PS; 4. RVH; 5. 1 PBF; 6. Olegimic lungs;
7. Mixing at aortic level.
Hemodynamics: Severity of TOF primarily depends on the severity of PS and extent of aortic overriding. Important hemodynamic changes in TOF are discussed below and presented in Fig. 17.18.
• PS is the most critical determinant of severity in ToF, presenting with absent P2 and ESM at PA. Although intensity of ESM increases with increasing severity of PS, it may be very soft in tight PS.
• RV outflow obstruction due to PS leads to—(a) increased RV pressure and (b) decreased PBF. However since RV has sufficiently large outlet through VSD and overriding aorta, CCF is rare in TOF despite PS.
• Increased RV pressure leads to Rt gt; Lt shunt through VSD. However, due to perimembranous location of VSD and overriding aorta, most of the shunt-flow enters directly into aorta and mixing of oxygenated and unoxygenated blood occurs at aortic level and not in the left ventricle. Magnitude of shunt at aortic level (and consequently the severity of cyanosis) depends on the extent of aortic overriding and RV pressure. An ejection click may be audible at aortic area due to dilatation of aortic root in some cases. Rt gt; Lt shunt at ventricular level via VSD is usually insignificant and silent, except in severe PS, due to lack of significant pressure gradient between two ventricles.
• Decreased PBF further contributes to cyanosis. However, in newborns or older cases with PDA, PBF may be maintained via Lt gt; Rt shunt at ductal level. In long-standing cases, major aortopulmonary collateral arteries (MAPCAs) may develop to augment PBF, producing functional flow murmurs over chest and back.
Clinical manifestations of TOF include:
• Cyanosis: Age of the appearance of cyanosis and its severity depends on the severity of PS. Most cases manifest by 8-12 weeks of life. Cyanosis is never present
at birth in TOF as relatively patent ductus during first few days of life allows enough flow to reach pulmonary circulation. Children with mild PS have insignificant Rt gt; Lt shunt and cyanosis may absent (Pink Fallot).
• Cyanotic or anoxic spells: Cyanotic spells are characterized by sudden, paroxysmal worsening of basal cyanosis with dyspnea, altered sensorium and occasionally seizures, lasting for few minutes. Exact etiology of these spells is unclear, probably relates to sudden and unexplained subpulmonic infundibular spasms, further diminishing the PBF. These attacks may occur at rest or in crying child with variable frequency, i.e. a few to multiple attacks every day. Cyanotic spells are more common lt;2 years of age and in anemic children.
• Squatting position: These children develop a preference for squatting or curled-up position by late infancy, as an adoptive mechanism to reduce systemic venous return (RV pressure) and increase systemic vascular resistance (LV pressure), thus reducing the Rt gt; Lt shunt.
• Clubbing: Although exact pathogenesis is unclear, chronic nail-bed hypoxia in cyanotic CHDs leads to dilatation of underlying capillaries, opening up of alternate AV fistulas, interstitial edema and reactive tissue hypertrophy, all leading to clubbing.
• Dyspnea of variable severity with effort intolerance or feeding difficulties is often the first manifestation, even before appearance of cyanosis.
• Growth retardation and failure to thrive, due to reduced oxygen supply and higher metabolic requirements is common.
Important signs in TOF are summarized in Table 17.20.
Complications include:
• Thromboembolic complications, due to compensatory polycythemia, e.g. (a) cerebral thrombosis, and (b) cerebral abscesses. Cerebral thrombosis is common lt; 2 years of age, while cerebral abscess is common in older children. Clinically, these cases present like cerebrovascular strokes.
| TABLE 17.20: Important signs in TOF | |
| Cyanosis*, clubbing, growth failure | |
| Inspection | Lateral shift of apex beat (RVH)* |
| Palpation | Left parasternal heave (RVH) |
| Auscultation | ESM at pulmonary area due to PS* Absent P2 (single S2)* Ejection click at aortic area Continuous flow murmurs (collaterals) |
| X-ray | Coure-en-sabot appearance* |
| ECG | RVH |
*characteristic clinical features
• Iron deficiency anemia, due to compensatory polycythemia,
• Metabolic complications, due to increased cell mass (polycythemia) coupled with hypoxic tissue injury, e.g. (i) xanthinuria and gout, (ii) hyperkalemia, (iii) hyperphosphatemia, (iv) hypocalcemia due to hyperphosphatemia, etc.
Infective endocarditis and CCF are rare in TOF.
Diagnosis is based on:
• Chest X-ray showing Cour en sabot appearance or bootshaped heart (Fig. 17.9.1) due to RVH with absent pulmonary conus and oligemic lung fields. Rightsided aortic arch is present in 25% cases.
• ECG showing RVH with right axis deviation.
• Echocardiography to—(a) establish exact location and severity of various lesions, (b) size of pulmonary vessels before surgery, (c) presence of associated abnormalities, e.g. anomalous origin of coronary vessels.
Doppler studies help to assess the pressure gradients between various chambers, specially across the pulmonary valve.• Cardiac catheterization is essential before surgery for precise assessment of structural defects and hemodynamic status. Selective right ventriculography or aortography may also be needed in some cases.
Management of ToF may be divided into—(a) medical management of cyanotic spells, (b) prevention and treatment of complications, (c) palliative measures to increase PBF, and (d) definite surgical correction.
a. Management of cyanotic spells includes:
• Knee-chest position to increase peripheral vascular resistance (and thus LV pressure) and decrease systemic venous return (and thus RV pressure), which helps to reduce Rt gt; Lt shunt.
• Humidified oxygen support, to improve oxygenation;
• IV fluid bolus of Normal saline 10-20 ml/kg;
• SC/IV morphine 0.1-0.2 mg/kg for sedation, to relieve anxiety and metabolic demands.
• #946;-blockers, e.g. IV Metoprolol (0.1 mg/kg over 5 min) to relieve infundibular spasm and reduce heart rate that improves ventricular filling. It can be repeated every 5 min if no hypotension or bradycardia, IV Esmolol infusion (50-200 mg/kg/min) may also be used in these cases.
• Correction of metabolic acidosis by IV sodium bicarbonate (1-2 mEq/kg) therapy, as required.
• Peripheral vasoconstrictors, e.g. IV/IM Methoxamine 0.1-0.2 mg/kg/dose to increase peripheral vascular resistance and LV pressure, thus to decrease Rt gt; Lt shunt.
• Refractory cases may need IV Phenylephrine infusion (2-5 #956;g#8725;kg#8725;min), IV Ketamine bolus (0.25-1.0 mg/ kg) or rarely, general anesthesia.
b. Prevention of complications includes:
• Cyanotic spells by continuous propranolol therapy (PO 0.5-1.0 mg/kg/dose qds)
• Thromboembolic complications by avoidance of dehydration. Severe polycythemia may need repeated phlebotomy or plasmapheresis to maintain hematocrit between 45-50%.
• Anemia by regular iron supplementation.
c. Palliative measures to increase PBF include:
• IV prostaglandin E1 infusion (0.05-0.2 mg/kg/min) may be used in newborns with severe cyanosis, as a temporary measure to keep the ductus arteriosus patent, till surgery is arranged.
• Palliative surgery is indicated in young and sick infants below 4 months with recurrent cyanotic spells, unlikely to withstand definitive correction. It aims to create an aortopulmonary communication (akin to ductus arteriosus), to augment the PBF. Most commonly used palliative surgery is Blalock-Taussig shunt with or without Gortex conduit between ipsilateral subclavian and pulmonary artery. Other approaches, e.g. Potts shunt between left pulmonary artery and descending aorta or Waterson shunt between right pulmonary artery and ascending aorta are rarely used. Presence of near-normal size pulmonary artery is essential for palliative surgery.
d. Definitive repair is indicated as a primary procedure in stable, minimally cyanosed cases or after palliative surgery at 6-12 months of age, using Broke procedure, i.e. closure of VSD and reconstruction of pulmonary valve.
Patient with absent pulmonary valve, if stable, must be managed medically till 1 year of age followed by total correction with repair of pulmonary artery branch dilation/aneurysm.
Operative mortality is ~2-5% and important postsurgical complications include arrhythmia, heart blocks and residual VSD or PS. Surgery is contraindicated in cases with hypertensive MAPCAS and severe ventricular dysfunction. Life-long infective endocarditis prophylaxis is indicated in operated cases.
2. Transposition of Great Arteries (TGA)
Transposition of great vessels/arteries (TGA) is the second commonest cyanotic CHD, characterized by reversal of the ventricular origin of great vessels, i.e. aorta arising from the RV and pulmonary artery from the LV.
Hemodynamically in TGA, systemic and pulmonary circulation run parallel to each other and survival depends on presence of some communication between two sides, e.g. ASD, VSD or PDA, to allow mixing of oxygenated and unoxygenated blood. PS may/may not be present.
Anatomically, TGA may be complete (as discussed above) or corrected in which, atrioventricular drainage is also reversed with RA draining in LV and LA in RV. Corrected TGA is usually asymptomatic unless other malformations are present, as the serial circulation between systemic and pulmonary flow is re-established. Clinical features of complete TGA depend on the presence or absence of VSD and PS.
• TGA without VSD/PDA manifest soon after birth with cyanosis and CCF.
• TGA with VSD/PDA but without significant PS usually present by 6-10 weeks with CCF.
• TGA with VSD/PDA as well as significant PS present usually by 6-10 weeks with cyanosis.
Auscultatory findings vary according to the type of lesion and have limited diagnostic value.
Diagnosis is supported by:
• Chest X-ray showing egg on string appearance, i.e. RVH with narrow base due to anteroposterior location of great vessels. Lung fields may be plethoric, normal or oligemic, depending on the severity of PS.
• ECG showing RVH with right-axis deviation, though biventricular hypertrophy or LVH may be present in cases with large VSD.
• Echocardiography is diagnostic, though cardiac catheterization is essential before surgery for precise assessment of the severity of lesions.
Management: Complete TGA needs early surgical intervention after stabilization of the patient and control of CCF. Prostaglandin E1 infusion is advised in severely cyanosed newborns to keep the ductus patent, till surgery is arranged.
Simple TGA or TGA with CoA should operated at 2-4 weeks or earlier if symptomatic. TGA with large VSDs must be operated by 6-8 weeks due to risk of early pulmonary vascular disease.
• Palliative Rashkind balloon atrial septostomy is indicated in very young and sick infants, to create an additional site (ASD) for mixing of systemic and pulmonary flow.
• Definite repair in younger infants involves Jatene arterial switch operation, i.e. resection of aortic and pulmonary trunk and re-anastamosis of distal aorta with proximal pulmonary trunk and vice versa, along with closure of VSD and bending of pulmonary artery. Other atrial-switch procedures, e.g. mustard or senning operation may be used in older children with small regressed LV but carry significant longterm morbidity.
3. Total Anomalous Pulmonary Venous
Drainage (TAPVC)
Total/partial anomalous pulmonary venous drainage (TAPVD / PAPVD) is an uncommon cyanotic CHD, characterized by anomalous drainage of all or some pulmonary veins into RA, superior vena cava or even portal vein, instead of LA.
Hemodynamically in these cases, mixing of oxygenated and unoxygenated blood occurs in RA, while LA receives blood only or partially through fossa ovalis/ASD. Increased blood flow in RA and then to RV, leads to RVH. PS is commonly present.
Clinical presentation depends on the extent of anomalous drainage and presence/severity of PS. Cases with significant PS usually manifest with cyanosis in second week of life, while those without PS manifest with CCF after neonatal period. Auscultatory findings are similar to ASD, i.e. wide and fixed split of S2 and ESM at pulmonary area, along with a venous hum in suprasternal or left infraclavicular region due to abnormal pulmonary veins.
Diagnosis rests on:
• X-ray chest showing 'Snowman in storm' or 'Figure of 8 appearance', due to abnormal position and dilatation of pulmonary veins with RVH.
• ECG shows RVH and right-axis deviation, usually associated with 'P' pulmonale suggestive of RA dilatation and rsR' pattern suggestive of RV strain in right chest leads.
• Echocardiography is diagnostic, though cardiac catheterization is essential before surgery for precisely assessment of the severity of lesions.
Management: Apart from supportive medical therapy (as for other cyanotic CHDs), treatment is essentially surgical. Surgery is indicated as early as possible in cases with TAPVD, as unoperated cases are unlikely to survive beyond 3 months. However, cases with PAPVD may survive for many years.
4. Tricuspid Atresia (TA)
Tricuspid atresia (TA) is a rare cyanotic CHD, characterized by congenital absence of tricuspid valve.
Hemodynamically, RA outflow obstruction leads to gross RA dilatation with Rt gt; Lt shunt via fossa ovalis or ASD, which is essential for survival. Additional flow in LA and LV leads to LVH. RV is hypoplastic and pulmonary circulation is sustained by a Lt gt; Rt shunt via VSD/PDA. PS is common.
Clinical presentation depends on the adequacy of Ltgt;Rt shunt at ventricular/ductal level. Most cases present at birth with cyanosis, LVH, pansystolic murmur of VSD, single S2 due to PS and pathognomonic prominent 'a' waves in jugular veins (due to RA contraction against atretic tricuspid valve).
Diagnosis is supported by—(a) X-ray chest suggestive of marked RA dilatation, LVH and oligemic lung fields, and (b) ECG showing LVH, left-axis deviation and 'P' pulmonale. It needs to be confirmed on echocardiography and/or cardiac catheterization.
Treatment includes a palliative Blalock-Taussig shunt in neonatal period, followed by modified Fontan operation at a later date, i.e. anastomosis of IVC and PA to bypass right heart. Prostaglandin E1 infusion is advised in severely cyanosed newborns to keep the ductus patent, till surgery is arranged.
5. Ebstein Anomaly (EA)
Ebstein anomaly is characterized by downward displacement of abnormal tricuspid valve into the right ventricle, i.e. atrialization of right ventricular cavity above the valve and consequent obstruction in RA outflow.
Hemodynamically, obstruction of the RA outflow tract leads to dilatation of RA and Rt gt; Lt shunt via fossa ovalis or ASD. Like tricuspid atresia, pulmonary flow in Ebstein anomaly is also largely dependent on VSD or PDA.
Clinically, these cases present at any age with exertional dyspnea, cyanosis or recurrent paroxysmal tachycardia. Important auscultatory findings include a typical scratchy murmur resembling pericardial rub in left parasternal area due to turbulence at tricuspid valve, with/without a PSM of associated VSD.
Diagnosis is supported by—(a) chest skiagram showing marked RA dilatation and LVH, and (b) ECG showing LVH, 'P' pulmonale due to RA abnormality and 'P' mitrale due to LA volume overload. It needs to be confirmed on echocardiography. Cardiac catheterization is necessary before surgery.
Management depends on the severity of defect. Surgical therapy aims to provide palliative treatment in newborns with severe cyanosis, i.e. closure of tricuspid valve by a synthetic patch, atrial septostomy and aortopulmonary shunt (Stranes procedure), followed by definitive valvuloplasty at a later date. Milder cases may be left untreated or taken directly for valvuloplasty in late childhood.
17.5.4
More on the topic CYANOTIC CHDs:
- Hyposplenic states
- RENAL VEIN THROMBOSIS
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025