CONGENITAL LUNG MALFORMATIONS
Congenital lung malformations are rare but important causes of respiratory morbidity in children with outcome dependent on the size of lesion and secondary changes,
e. g. infection or hemorrhage.
Failure to recognize them may lead to inappropriate delay in appropriate management.Pulmonary agenesis/aplasia denotes complete absence of a lung. Bronchial stump is also absent in agenesis but present in aplasia. Pulmonary agenesis may be autosomal recessive or associated with other malformations, e.g. VACTERL sequence.
Clinically, bilateral lung agenesis is incompatible with life, while unilateral defects may be asymptomatic due to compensatory growth of the remaining lung or present at any age with non-specific respiratory illness. Scoliosis is common in older children.
Chest skiagram shows unilateral lung or lobar collapse and diagnosis must be confirmed by CT/MRI. Other congenital anomalies must also be excluded.
Treatment is conservative, though surgery may benefit selected cases.
Pulmonary hypoplasia denotes decrease in numbers of alveoli and airway generations, usually due to intrauterine pressure that prevents normal lung development, e.g. diaphragmatic hernia or oligohydramnios. Depending on the severity, pulmonary hypoplasia may present in newborns with respiratory insufficiency or persistent pulmonary hypertension or at a later age with recurrent respiratory illnesses. Diagnosis requires lung imaging studies, preferably MRI. Treatment depends on the severity and some cases may need mechanical ventilation, treatment of pulmonary hypertension or extracorporeal membrane oxygenation to sustain life.
Congenital cystic adenomatous malformation (CCAM) or congenital pulmonary airway malformation (CPAM) is the presence of hamartomatous cystic lung tissue mixed with normal lung due to mal-development of terminal bronchiolar structures in early gestation.
Among the five histologic types, commonest is type I disease (60%) with single or multiple large cysts involving only a part of one lung.Clinically, most cases present in early infancy with respiratory distress, recurrent respiratory infections and pneumothorax, often confused with a diaphragmatic hernia or lung abscess.
Diagnosis rests on CT scan (Fig. 16.15), and prenatal diagnosis is possible on ultrasonography. Treatment is surgical, though may be delayed in asymptomatic cases
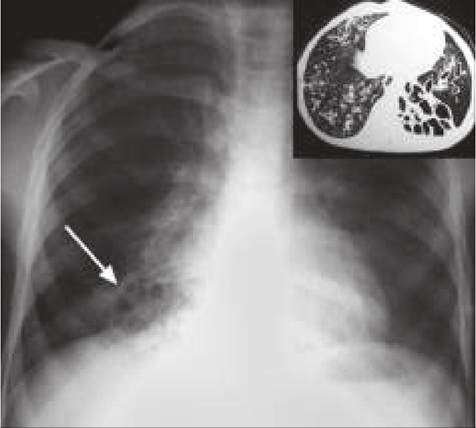
Fig. 16.15: Congenital cystic adenomatous malformation.
till one year of age due to rare possibility of spontaneous resolution. Malignant changes are possible in unoperated cases.
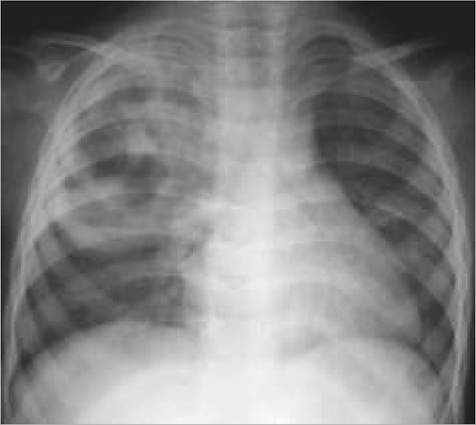
Fig. 16.16: Bronchogenic cyst.
Antenatal intervention is controversial as many pre- natally detected cysts regress in size postnatally.
Pulmonary sequestration is a congenital anomaly of lung development in which a part of the lung tissue does not connect to a bronchus and receives its blood supply from the systemic arteries. Sequestration may be Intrapulmonary (more common) or extrapulmonary, depending on the location. Associated anomalies, e.g. CCAM and diaphragmatic hernia are not uncommon.
Clinically, sequestration functions as a space occupying lesion in the chest and most cases are asymptomatic. Some cases present with secondary infection, abscess or hemoptysis.
Diagnosis is confirmed by CT chest and contrast studies are necessary to assess vascular supply before surgery.
Treatment is surgical though coil embolization of feeding artery may be successful in selected cases.
Bronchogenic cysts denote abnormal budding of the tracheal diverticulum of the foregut, more common on the right, near midline. Rarely demonstrable at birth, these cysts usually present later after secondary infection and enlargement. Large cyst may also compress adjacent airway.
Chest skiagram usually shows solid coin lesions or air-filled cyst with/without fluid level (Fig. 16.16), though diagnosis must be confirmed on CT/MRI before surgery.
Treatment is surgical excision after appropriate antibiotics. Excision is advised even for asymptomatic cysts due to risk of future infection.
16.11