Dystrophinopathies
Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is an X-linked disorder caused by an abnormality at the Xp21 gene loci. The DMD/BMD gene occupies 2.5 million base pairs of DNA on the X chromosome and is about 10 times larger than the next largest gene identified to date.
The gene coding sequence contains 79 exons. The primary protein product, dystrophin, is localized to the intracellular side of the plasma membrane of all myogenic cells, certain types of neurons, and in small amounts of other cell types (5). Dystrophin deficiency at the plasma membrane of muscle fibers disrupts the membrane cytoskeleton and leads to the secondary loss of other components of the muscle cytoskeleton. The primary consequence of the cytoskeleton abnormalities is membrane instability, leading to membrane injury from mechanical stresses, transient breaches of the membrane, and membrane leakage. Chronic dystrophic myopathy is characterized by aggressive fibrotic replacement of the muscle and eventual failure of regeneration with muscle fiber death and fiber loss. Generally, loss of the reading frame causes complete absence of dystrophin and a Duchenne phenotype. For cases with a deletion mutation, the “reading frame” hypothesis predicts that BMD patients with inframe deletions produce a semifunctional, internally deleted dystrophin protein, whereas DMD patients with frameshift or “out of frame” deletions produce a severely truncated protein that would be unstable (6). Characteristics of DMD and BMD are shown in Table 12.1.Diagnostic Evaluation. Serum creatine kinase is a useful screening test. Gene abnormalities may be identified by full gene sequencing of a blood specimen in 99% of all patients with a dystrophinopathy. Full gene sequencing in addition to evaluation for large deletions to identify point mutations, deletions, duplications, inversions, etc.
rather than simple deletion screening with polymerase chain reaction (PCR) is now standard of care in all patients at risk of a dystrophinopathy. This is both for diagnostic purposes and to identify candidates for future molecular-based therapies such as exon skipping with oligonucleotides, nonsense- mediated suppression therapy for the 10% to 15% of patients with DMD and BMD with stop codon mutations, and specific gene therapy strategies that will require knowledge of specific gene sequence alterations. In patients with no family history and molecular genetics that do not clearly differentiate a DMD and BMD phenotype, a muscle biopsy with immunostaining and quantitative dystrophin analysis with Western blot is critical to allow patients to be eligible for future clinical trials with rigid inclusionary criteria.12.1
Characteristics of Dystrophinopathies
| DUCHENNE MUSCULAR DYSTROPHY | BECKER MUSCULAR DYSTROPHY | |
| U.S. Prevalence (est.) | 15,000 | 2,200 |
| Incidence rate | 1/3,500 male births | unknown |
| Inheritance | X-linked | X-linked |
| Gene location | Xp21 (reading frame shifted) | Xp21 (reading frame maintained) |
| Protein | Dystrophin | Dystrophin |
| Onset | 2 to 6 years | 4-12 years (severe BMD) Late teenage to adulthood (mild BMD) |
| Severity and course | Relentlessly progressive Reduced motor function by 2-3 yrs Steady decline in strength Life span 16 years | |
| Weakness | Proximal > distal Symmetric Legs and arms | Proximal > distal Symmetric Legs and arms |
| Cardiac | Dilated cardiomyopathy first to second decade Onset of signs second decade | Cardiomyopathy (may occur before weakness); third to fourth decade frequent |
| Respiratory | Profoundly reduced vital capacity in second decade Ventilatory dependency in second decade | Respiratory involvement in subset of patients Ventilatory dependency in severe patients |
| Muscle size | Calf hypertrophy | Calf hypertrophy |
| Musculoskeletal | Contractures: ankles, hips, and knees Scoliosis: onset after loss of ambulation | Contractures: ankles and others in adulthood |
| CNS | Reduced cognitive ability oReduced verbal ability | Some patients have reduced cognitive ability |
| Muscle pathology | Endomysial fibrosis and fatty infiltration Variable fiber size and myopathic grouping Fiber degeneration/regeneration Dystrophin: absent Sarcoglycans: secondary reduction | Variable fiber size Endomysial connective tissue and fatty infiltration Fiber degeneration Fiber regeneration Dystrophin: reduced (usually 10%-60% of normal) |
| Blood chemistry and hematology | CK: Very high (10,000-50,000) High AST and ALT (normal GGT) High aldolase | CK: 5,000 to 20,000 Lower levels with increasing age |
Epidemiology.
The incidence of Duchenne muscular dystrophy, based on a number of population studies as well as neonatal screening, has been estimated to be around 1:3,500 male births (7). As many as one-third of isolated cases may be due to new mutations, which is considerably higher than observed in other X-linked conditions. This high mutation rate may relate to the large size of the gene.Onset and Early Signs. While the history of hypotonia and delayed motor milestones are often reported in retrospect, the parents are often unaware of any abnormality until the child starts walking. Variability has been reported in the age of onset (8,9). In 74% to 80% of instances, the onset has been noted before the age of 4 years (8-10). The vast majority of cases are identified by 5 to 6 years of age. The most frequent presenting symptoms have been abnormal gait, frequent falls, and difficulty climbing steps. Parents frequently note the toe walking, which is a compensatory adaptation to knee extensor weakness, and a lordotic posture to the lumbar spine, which is a compensatory change due to hip extensor weakness (Fig. 12.7).
Occasionally, Duchenne muscular dystrophy is identified presymptomatically in situations where a CK value is obtained with a markedly elevated value, malignant hyperthermia occurs during general anesthesia for an unrelated surgical indication, or a diagnosis is pursued in a male with an affected older sibling.
Difficulty negotiating steps is an early feature, as is a tendency to fall due to the child tripping or stumbling on a plantar-flexed ankle or the knee buckling or giving way due to knee extensor weakness. There is progressive difficulty getting up from the floor with presence of a Gower's sign (see Fig. 12.5).
Pain in the muscles, especially the calves, is a common symptom. Enlargement of muscles, particularly the calves (see Fig. 12.1), is commonly noted. The deltoid may also be hypertrophied. With the patients arms abducted to 90 degrees and externally rotated, the hypertrophy of the posterior deltoid and infraspinatus frequently leaves a depression between these two muscles referred to as the “posterior axillary depression sign” in DMD (Fig.
12.9). The tongue is also frequently enlarged. There is also commonly an associated wide arch to the mandible and maxilla with separation of the teeth, presumably secondary to the macroglossia.Pattern and Progression of Weakness. Earliest weakness is seen in the neck flexors during preschool years (Fig. 12.10). Weakness is generalized, but predominantly proximal early in the disease course. Pelvic girdle weakness predates shoulder girdle weakness by several years. Ankle dorsiflexors are weaker than ankle plantar flexors; ankle everters are weaker than ankle inverters; knee extensors are weaker than knee flexors; hip extensors are weaker than hip flexors; and hip abductors are weaker than hip adductors (9).
The weakness progresses steadily, but the rate may be variable during the disease course. Quantitative strength testing shows greater than 40% to 50% loss of strength by 6 years of age (9). With manual muscle testing, DMD subjects exhibit loss of strength in a fairly linear fashion from ages 5 to 13, and measurements obtained several years apart will show fairly steady disease progression. A variable course may be noted when analyzing individuals over a shorter time course (9). Quantitative strength measures have been shown to be more sensitive for demonstrating strength loss than manual muscle testing when strength is grades 4-5 (9).
Loss of Ambulation. Average age to wheelchair in a DMD population not treated with corticosteroids has
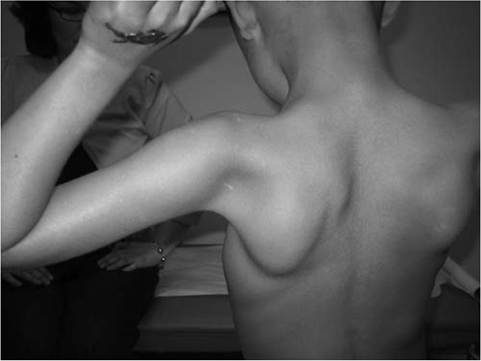
Figure 12.9 Posterior axillary depression sign in Duchenne muscular dystrophy. Note the prominent deltoid superolaterally and infraspinatus inferomedially.
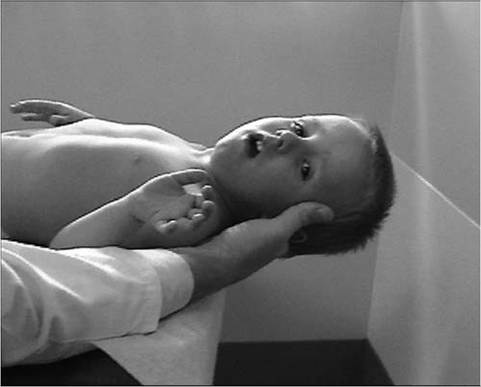
Figure 12.10 Weakness of neck flexors in an eight-year-old child with Duchenne muscular dystrophy makes it difficult for him to bring his chin to the chest when supine and to hold his head up when placed at the end of the examination table.
been age 10, with a range of 7-13 years. Treatment with prednisone or deflazacort helps maintain strength and prolongs ambulation by two years (11,12). There does not appear to be an advantage of deflazacort over daily prednisone. The optimal dose of prednisone is 0.75 mg/ kg/day up to a maximum of 40 mg/day (11,12,13). The optimal dose of deflazacort appears to be 0.90 mg/kg/ day. With both corticosteroid regimens, patients need to be monitored for cataracts, hypertension, weight gain, osteoporosis, growth retardation, diabetes, and behavioral side effects.
Timed motor performance is useful for the prediction of time when ambulation will be lost without provision of long-leg braces. One large natural history study showed that all DMD subjects who took nine seconds or longer to ambulate 30 feet lost ambulation within two years. All DMD subjects who took 12 seconds or longer to ambulate 30 feet lost ambulation within one year (9). Ambulation past the age of 14 in a noncorticosteroid-treated patient should raise the suspicion of a milder form of muscular dystrophy such as BMD or limb girdle muscular dystrophy. Ambulation beyond 16 years has been previously used as an exclusionary criteria for Duchenne muscular dystrophy in studies of BMD. Immobilization for any reason can lead to a marked and often precipitous decline in muscle power, rapid development of contractures, and loss of ambulatory ability. A fall with resultant fracture leading to immobilization and loss of ambulatory ability is not an uncommon occurrence.
Contractures. Significant joint contractures have been found in nearly all children with Duchenne muscular dystrophy older than age 13 (9,14,15). The most common contractures include ankle plantar flexion, knee flexion, hip flexion, iliotibial band, elbow flexion, and wrist flexion contractures (9). Significant contractures have been shown to be rare in DMD before age 9 for all joints. There is no association between muscle imbalance around a specific joint (defined as grade 1 or greater difference in flexor and extensor strength) and the frequency or severity of contractures involving the hip, knee, ankle, wrist, and elbow in DMD (9).
Flexion contractures have been shown to be rare in those with ≥grade 3 extensor strength about a joint, an expected finding because of the definition of a grade 3 muscle on manual muscle testing (MMT). For those DMD subjects with less than antigravity strength about a joint, there is low correlation between the MMT strength of these specific muscle groups and the severity of joint contracture (9). The presence of lower extremity contractures in DMD has been shown to be strongly related to onset of wheelchair reliance (9). Lower extremity contractures were rare while DMD subjects were still upright, but developed soon after they developed a sitting position in a wheelchair for most of the day. The occurrence of elbow flexion contractures also appears to be directly related to prolonged static positioning of the limb, and these contractures develop soon after wheelchair reliance. The relationship between wheelchair reliance and hip and knee flexion contractures has been noted (9). Mild contractures of the iliotibial bands, hip flexor muscles, and heel cords occurs in most DMD patients by 6 years of age (16). Limitations of knee, elbow, and wrist extension occurs about two years later (9,16); however, these early observed contractures were relatively mild. Given the tremendous replacement of muscle by fibrotic tissue in DMD subjects, it is not surprising that a muscle of less than antigravity extension strength, statically positioned in flexion, would develop a flexion contracture (subsequent to wheelchair reliance). The lack of lower extremity weight bearing likely contributes to the rapid acceleration in the severity of these contractures after transition to wheelchair. Ankle plantar flexion contractures are not likely a significant cause of wheelchair reliance, as few subjects exhibit plantar flexion contractures of ≥15 degrees before their transition to a wheelchair (9). Natural history data suggests that weakness is the major cause of loss of ambulation in DMD, not contracture formation.Spine Deformity. Reported ultimate prevalence of scoliosis in DMD subjects not treated with corticosteroids varies from 33% to 100% (17). This marked variability is primarily because of retrospective selection for scoliosis, the inclusion or exclusion of functional curves, and dissimilar age groups. The prevalence of scoliosis is strongly related to age. Fifty percent of DMD patients acquire scoliosis between ages 12 and 15, corresponding to the adolescent growth spurt. Ten percent of older DMD subjects with no treatment of scoliosis show no clinical spinal deformity. This is consistent with Oda's report (18) that 15% of older DMD patients show mild nonprogressive curves (usually 10 degrees to 30 degrees). The rate of progression of the primary or single untreated lateral curve has been reported to range from 11 degrees to 42 degrees per year, depending on the age span studied. Johnson and Yarnell (19) reported an association between side of curvature, convexity, and hand dominance, an association recently confirmed (20). Oda and colleagues (18) reported that the likelihood of severe progressive spinal deformity could be predicted by type of curve and early pulmonary function measurements. Those with spines lacking significant kyphosis or hyperlordosis and a peak obtained absolute forced vital capacity (FVC) greater than 2,000 mL tended not to show severe progressive scoliosis.
No cause-and-effect relationship has been established between onset of wheelchair reliance and occurrence of scoliosis (9,21). Wheelchair reliance and scoliosis has been found to be an age-related phenomenon. The causal relationship between loss of ambulatory status and scoliosis is doubtful, given the substantial time interval between the two variables in most subjects (scoliosis usually develops after three to four years in a wheelchair). Both wheelchair reliance and spinal deformity may be significantly related to other factors (eg, age, adolescent growth spurt, increase in weakness of trunk musculature, and other unidentified factors) and thus represent coincidental signs of disease progression.
In retrospective series, treatment of DMD with deflazacort and prednisone have been shown to reduce the occurrence of significant scoliosis (22,23,24). It remains to be seen whether the apparent arrest in the development of scoliosis with corticosteroids will continue past the age of skeletal maturity.
Pulmonary Function. In DMD, absolute forced vital capacity volumes increase during the first decade and plateau during the early part of the second decade (9). A linear decline in percent predicted FVC is apparent between 10 and 20 years of age in DMD (9). Rideau and colleagues (25) reported forced vital capacity to be predictive of the risk of rapid scoliosis progression. In the most severe DMD cases, maximal forced vital capacity reached a plateau of less than 1,200 mL. This was associated with loss of ability to walk before age 10 and severe progressive scoliosis. Moderately severe DMD cases with respiratory compromise reached maximum forced vital capacities between 1,200 mL and 1,700 mL. Spinal deformity was present consistently in these cases, but varied in severity. The least severe DMD cases reached plateaus in FVC of greater than 1,700 mL. Similarly, McDonald and colleagues (9) found that those patients with higher peak FVC (>2,500 mL) had a milder disease progression, losing 4% predicted FVC per year. Those with peak predicted FVC less than 1,700 mL lost 9.6% predicted FVC per year. Thus, the peak obtained absolute values of forced vital capacity usually occurring in the early part of the second decade is an important prognostic indicator for severity of spinal deformity, as well as ultimate severity of restrictive pulmonary compromise due to muscular weakness. Prednisone and deflazacort both appear to reduce the loss of pulmonary function over time during the second decade in DMD (22,23,24,26).
Maximal static airway pressures (both maximal inspiratory pressure and maximal expiratory pressure) are the earliest indicators of restrictive pulmonary compromise in DMD with impaired values noted between 5 and 10 years of age. Vital capacity typically increases concomitant with growth between 5 and 10 years of age, with percent predicted FVC remaining relatively stable and close to 100% predicted. DMD patients typically show a linear decline in percent predicted FVC between 10 and 20 years of age. An FVC falling below 35% is associated with increased perioperative morbidity in DMD (27) and optimally, surgery should ideally be performed with % predicted FVC greater than 40%. Recent evidence suggests that spinal arthrodesis may be safely performed in a population of DMD with % predicted vital capacity less than 30% (28).
Ultimately, respiratory failure in DMD is insidious in its onset and results from a number of factors, including respiratory muscle weakness and fatigue, alteration in respiratory system mechanics, and impairment of the central control of respiration. Noninvasive forms of both positive and negative pressure ventilatory support are increasingly being offered to DMD patients nocturnally and continuously with acceptable quality of life. Airway clearance strategies, such as the cough assist/inexsufflator, TheraVest, or intrapulmo- nary percussion ventilation (IPV) are also important pulmonary management strategies (29).
Cardiomyopathy. The dystrophin protein is present in both the myocardium and the cardiac Purkinje fibers. Abnormalities of the heart may be detected by clinical examination, electrocardiogram (ECG), echocardiography, and Holter monitoring. Cardiac examination is notable for the point of maximal impulse palpable at the left sternal border due to the marked reduction in anteroposterior chest dimension common in DMD. A loud pulmonic component of the second heart sound suggests pulmonary hypertension in patients with restrictive pulmonary compromise. Nearly all patients over the age of 13 demonstrate abnormalities of the ECG (9). Q-waves in the lateral leads are the first abnormalities to appear, followed by elevated ST segments and poor R-wave progression, increased R/S ratio, and finally resting tachycardia and conduction defects. ECG abnormalities have been demonstrated to be predictive for death from the cardiomyopathy with the major determinants including R-wave in lead V1 less than 0.6 mV; R-wave in lead V5 less than 1.1 mV; R-wave in lead V6 less than 1.0 mV; abnormal T-waves in leads II, III, AVF, V5, and V6; cardiac conduction disturbances; premature ventricular contraction; and sinus tachycardia (30). Sinus tachycardia may be due to low stroke volume from the progressive cardiomyopathy, or in some cases, may be sudden in onset and labile, suggesting autonomic disturbance or direct involvement of the sinus node by the dystrophic process (31).
Autopsy studies and thallium 201 single-photon emission computed tomography (SPECT) imaging have demonstrated left ventricular lateral and posterior wall defects that may explain the lateral Q-waves and the increased R/S ratio in V1 seen on ECG. Localized posterior wall fibrosis was found to be peculiar to DMD and was not found in other types of muscular dystrophy (32). Pulmonary hypertension leading to right ventricular enlargement also is known to affect prominent R-waves in V1 and has been demonstrated in patients with DMD (33).
Ventricular ectopy and sudden death are known complications of the cardiomyopathy in DMD, and this association likely explains observed cases of sudden death. Severe ventricular ectopy in DMD has been associated with left ventricular dysfunction and sudden death. Yanagisawa and colleagues (34) reported an age-related increase in the prevalence of cardiac arrhythmias detected by ambulatory 24-hour electrocardiographic recordings. They also noted an association between ventricular arrhythmias and sudden death in DMD. Clinically evident cardiomyopathy is usually first noted after age 10 and is apparent in nearly all patients over age 18 (35). Development of cardiomyopathy is a predictor of poor prognosis. Echocardiography has been used extensively to follow the development of cardiomyopathy and predict prognosis in patients with DMD. The onset of systolic dysfunction noted by echocardiography is associated with a poor short-term prognosis (35). The myocardial impairment remains clinically silent until late in the course of the disease, possibly caused by the absence of exertional dyspnea, secondary to lack of physical activity. Death has been attributed to congestive heart failure in as many as 40% to 50% of patients with DMD by some investigators (35). Regular cardiac evaluations with an ECG, echocardiography, and Holter monitor should be employed in teenagers with preclin- ical cardiomyopathy.
Recent studies suggest that early presymptom- atic treatment to achieve afterload reduction with angiotensin-converting enzyme inhibitors (ACE inhibitors) such as perindopril or enalapril delayed the onset and progression of prominent left ventricle dysfunction in children with DMD (36). In another series, 43% with impaired left ventricular (LV) systolic dysfunction responded to enalapril with the normalization of function (37). Alternatively, angiotensin II type 1 receptor blockers (ARBs) such as losartan may be considered for afterload reduction in DMD. Animal studies show that the angiotensin II type 1 receptor blocker losartan attenuates TGF-beta-induced failure of muscle regeneration in dystrophinopathy, presenting an additional potential for therapeutic benefit vis-a-vis skeletal muscle in DMD (38).
Cognition and Behavioral Phenotype. There is a dystrophin isoform present in the brain. Previous studies on intellectual function on children with DMD have generally revealed decreased IQ scores when these children are compared with both control and normative groups (9). A mean score for the DMD population of 1.0 to 1.5 standard deviation (SD) below population norms has been reported. There has generally been a considerable consistency in the degree of impairment across measures reflecting a rather mild global deficit. Some studies (39) have demonstrated relative deficits in verbal IQ. In a longitudinal assessment of cognitive function, McDonald and colleagues (9) found IQ measure in DMD to be stable over time. On neuropsychological testing, a large proportion of DMD subjects fell within the “mildly impaired” or “impaired” range according to normative data (9). Again, no particular areas of strength or weakness were identified. These findings likely reflect a mild global deficit rather than focal nervous system impairment (9). Hinton found that DMD is associated with poor attention to complex verbal information (more so than verbal or memory measures), and they exhibit decreased verbal span capacity, but not impaired recall (40,41). An increased incidence of autism spectrum disorder has been found in DMD (42). In one large series of DMD subjects, 11.7% were reported to have a comorbid diagnosis of attentiondeficit hyperactivity disorder (ADHD), 3.1% had autism spectrum disorder, and 4.8% had obsessivecompulsive disorder (43). In addition, impaired facial affect recognition has found to be a part of the phenotype associated with DMD (44).
Anthropometric Changes. Substantial anthropometric alterations have been described in DMD. Short stature and slow linear growth with onset shortly after birth has been reported (45). Accurate measurement of linear height is extremely difficult in this population. Arm span measurements may be an alternative measure of linear growth; however, this measurement might also be difficult, as elbow flexion contractures of greater than 30 degrees are frequently present in patients older than age 13. Forearm segment has been proposed as an alternative linear measurement in DMD patients with proximal upper extremity contractures, and radius length may be followed for those with wrist and finger contractures. Obesity is a substantial problem in DMD, subsequent to the loss of independent ambulation (9,46). Weight control during early adolescence has its primary rationale in ease of care, in particular, ease of transfers during later adolescence.
Immediately following spine fusion, there has been a documented tendency for DMD patients to lose significant weight. Those who lost weight were unable to self-feed. The weight loss after surgery was associated with loss of self-feeding (47). There was no association with weight loss and loss of biceps strength. A correction of the kyphosis may actually make self-feeding problematic in DMD. A feeding evaluation and incorporation of kyphosis into the spinal instrumentation construct may help preserve self-feeding and prevent weight loss subsequent to spine fusion.
Longitudinal weight measurements in DMD confirm significant rates of weight loss in subjects ages 17-21 (9,48). This is likely caused by relative nutritional compromise during the later stages when boys with DMD have higher protein and energy intake requirements because of hypercatabolic protein metabolism. Protein and calorie requirements may often be 160% of that predicted for able-bodied populations during the later stages of DMD (49,50). Restrictive lung disease becomes more problematic during this time, and this may also influence caloric intake and requirements. Self-feeding often becomes impossible during this period because of significant biceps weakness. In addition, boys with DMD may develop signs and symptoms of upper gastrointestinal dysfunction (51).
Becker Muscular Dystrophy
Existence of a form of muscular dystrophy with a similar pattern of muscle weakness seen in Duchenne muscular dystrophy, X-linked inheritance, but with later onset and a much slower rate of progression, was first described by Becker and Kiener in 1955 (52). The disorder has the same gene location as the DMD gene (Xp21) and is thus allelic. On immunostaining of muscle biopsy specimens, the presence of patchy abundance of dystrophin suggests a Becker muscular dystrophy phenotype. On Western blot for quantitative dystrophin analysis, either 20% to 80% dystrophin levels or normal quantity and reduced or increased molecular-weight dystrophin is consistent with BMD. Studies show that 5% to 20% dystrophin quantity is consistent with an outlier or intermediate phenotype (5).
Epidemiology. Becker muscular dystrophy has a lower incidence than DMD, with prevalence rates for BMD ranging from 12-27 per million and a recent estimated overall prevalence of 24 per million (7,53).
Molecular Genetics and Diagnostic Evaluation. Full gene sequencing of the dystrophin gene, which demonstrates large deletions, duplications, and point mutations, identifies 99% of patients with dystrophinopathy and is now the standard of care. This is essential for identification of patients with stop codons and specific gene alterations that will be targeted for molecularbased therapies. Not all DMD and BMD patients have deletion mutations: Many have point mutations that cannot be detected by screening deletion testing. Thus, full sequence analysis is necessary. About 55% of DMD patients and 70% of BMD patients show large deletion mutations of the gene. A positive DNA test result (presence of a point mutation, duplication, or deletion) is diagnostic of a dystrophinopathy (Duchenne or Becker dystrophy)—there are no false-positives if the test is done appropriately. While genetic testing is improving with regard to the differentiation of DMD and BMD, there remains some overlap and variability. Differential diagnosis between DMD and BMD is best done by a consideration of clinical findings, family history of clinical phenotype, and muscle biopsy with quantitative dystrophin analysis. If the patient is still ambulating at 16-20 years of age and has a deletion mutation, the correct diagnosis is BMD. Mutations at the Xp21 locus, which maintain the translational reading frame (in-frame mutations), result in an abnormal but partially functional dystrophin protein, whereas in Duchenne muscular dystrophy, the mutations shift the reading frame (out-of-frame mutations) so that virtually no dystrophin is produced. The reading frame interpretation is most accurate for deletions in the center of the gene (exons 40-60) and is least accurate for deletions in the beginning of the gene (exons 1-20).
Absent dystrophin or levels less than 5% of normal generally are considered diagnostic of Duchenne muscular dystrophy; however, 5% of such patients have BMD phenotypes. In BMD, dystrophin typically has abnormally small molecular weight (427 kDa) or normal molecular weight. Most BMD patients with larger or smaller molecular-weight dystrophin also have decreased quantities of the protein. All BMD patients with normal molecular-weight dystrophin have decreased quantities, usually less than 30% normal. Smaller-size dystrophin typically is caused by deletion mutations, and larger-size dystrophin by duplication mutations. A further refinement is the use of antibodies specific to the carboxy-terminal (C-terminal) region of dystrophin. Using such antibodies, immunohistochemistry reveals that the C-terminal region is almost always absent in DMD but invariably present in BMD. Thus, when this region of the molecule is missing, a more severe phenotype is likely.
Age of Onset and Presenting Signs. Studies have shown significant overlap in the observed age of onset between DMD and BMD (10). Although determination of the quantity and molecular weight of dystrophin has substantially improved the early differentiation among BMD, “outlier” DMD, and the more common and rapidly progressive DMD phenotype, Bushby and colleagues (54) found no clear correlation between abundance of dystrophin and clinical course within the BMD group.
A series of Bushby and Gardner-Medwin (54), which included 67 BMD subjects, supported the presence of two major patterns of progression in BMD: a “typical” slowly progressive course and a more “severe” and rapidly progressive course. All of the “severe” BMD cases showed difficulty climbing stairs by age 20, whereas none of the “typical” BMD cases had difficulty climbing stairs before age 20. Abnormal ECGs were seen in 27% of typical BMD subjects and 88% of severe subjects. Bushby and Gardner-Medwin (54) found BMD subjects to have a mean age of onset of 12 years in the typical group and 7.7 years in the severe group. Some patients with BMD present with major muscle cramps as an isolated symptom (54). As in DMD, preclinical cases are often identified by the finding of a grossly elevated CK value. There is also considerable overlap in CK values between DMD and BMD cases at the time of presentation. Thus, CK values cannot be used to differentiate DMD from BMD.
Calf enlargement is a nonspecific finding in BMD, as is the presence of a Gower's sign. The gait over time is similar to other neuromuscular disease conditions with proximal weakness. Patients often ambulate with a lumbar lordosis, forefoot floor contact, decreased stance-phase knee flexion, and a Trendelenburg's or gluteus medius lurch, often described as a waddle.
Other atypical clinical presentations include a sole complaint of cramps on exercise in individuals with no muscle weakness (54). In addition, patients with focal wasting of the quadriceps, previously diagnosed with quadriceps myopathy, have been diagnosed with BMD, based on molecular genetic testing and/or dystrophin analysis on muscle biopsy (10).
Age of Transition to Wheelchair. The most useful clinical criterion to distinguish BMD from DMD is the continued ability of the patient to walk into late teenage years. Those with BMD will typically remain ambulatory beyond 16 years. Some patients may become wheelchair uses in their late teens or 20s, whereas others may continue walking into their 40s, 50s, or later. DMD cases usually stop ambulating by 13 years unless treated with corticosteroids. Outlier DMD or intermediate dystrophinopathy cases generally stop ambulating between 13 and 16 years of age.
Pattern and Progression of Weakness. BMD patients have distribution of weakness similar to those with DMD (10). Proximal lower limb muscles are involved earlier in the disease course. Gradual involvement of the pectoral girdle and upper limb musculature occurs 10-20 years from onset of disease. Extensors have been noted to be weaker than flexors (10). The muscle groups that are most severely involved earlier in the course of disease include the hip extensors, knee extensors, and neck flexors (10).
Contractures. Early development of contractures does not appear to be a feature of BMD (10,54). As with BMD, nonambulatory BMD subjects may develop equinus contractures, knee flexion contractures, and hip flexion contractures. Because of the tremendous replacement of muscle in BMD subjects by fibrotic tissue, it is likely that, as in DMD, a muscle with less- than-antigravity extension strength, which is statically positioned in flexion, is more likely to develop a flexion contracture subsequent to wheelchair reliance.
Spine Deformity. Spinal deformity is not nearly as common or severe in BMD, as compared with DMD. Spinal instrumentation is rarely required by DMD patients
(10.54).
Pulmonary Function. Compromised pulmonary function is much less problematic in BMD as opposed to DMD
(10.25.54). The percent predicted forced vital capacity does not appear substantially reduced until the third to fourth decade. The percent predicted maximal expiratory pressure appears relatively more reduced at younger ages than the percent predicted maximal inspiratory pressure, a finding seen in DMD and other neuromuscular diseases (9,55,56,57). This may be caused by more relative involvement of the intercos- tals and abdominal musculature with relative sparing of contractile function in the diaphragm of BMD. As in DMD and other neuromuscular disease, it appears that predicted maximal expiratory pressure (MEP) may be a useful quantitative measure of impairment and perhaps disease progression early in the course of BMD.
Cardiomyopathy. The pattern of occasional life-threatening cardiac involvement in otherwise mild and slowly progressive BMD has been reported by many (54,58). A significant percentage of BMD cases develop cardiac abnormalities, and the rate of progression of cardiac failure may on occasion be more rapid than the progression of skeletal myopathy (58). In fact, successful cardiac transplantation has been successfully performed in BMD subjects with cardiac failure. Approximately 75% of BMD patients have been found to exhibit ECG abnormalities (10,59). The abnormal findings most typically reported include abnormal Q-waves, right ventricular hypertrophy, left ventricular hypertrophy, right bundle branch block, and nonspecific T-wave abnormalities. Unlike DMD, resting sinus tachycardia has not been a frequent finding. Echocardiography has shown left ventricular dilation in 37%, whereas 63% have subnormal systolic function because of global hypokinesia (59). Thus, the cardiac compromise may be disproportionately severe, relative to the degree of restrictive lung disease in some BMD subjects. The evidence for significant myocardial involvement in BMD is sufficient to warrant screening of all of these patients at regular intervals using ECG and echocardiography. The slowly progressive nature of this dystrophic myopathy, which is compatible with many years of functional mobility and longevity, makes these patients suitable candidates for cardiac transplantation if end-stage cardiac failure occurs.
Some cases with BMD may present with an isolated cardiomyopathy with no clinical manifestation of skeletal muscle involvement. The diagnosis can be established by demonstration of a deletion in the Xp21 gene or by muscle biopsy. Isolated cases of cardiomyopathy in children, particularly those with family histories indicative of X-linked inheritance, should be screened for BMD with an initial serum CK estimation and molecular genetic studies of the Xp21 gene.
Cognition. Cognitive testing in BMD subjects have shown large variability in IQ scores and neuropsychological test measures. Mildly reduced intellectual performance has been noted in a subset of BMD patients; however, the degree of impairment is not as severe as noted in DMD (10).