Growth and developmental problems in children are common causes of parental concerns, though all are not necessarily pathological.
Samir H Dalwai, Surbhi Rathi, Mukesh Agrawal
This chapter aims to distinguish normal variations from pathological disorders, as well as to discuss diagnostic and therapeutic approach in some common problems of growth and development.
3.1 FAILURE TO THRIVE
Failure to thrive (FTT) is a clinical state of growth failure due to any cause, characterized by any one or more of the following features:
a. Lack of growth, i.e. weight for age < 3rd percentile
b. Failure to gain weight over time on serial monitoring
c. Deceleration in growth velocity, i.e. weight dropping at least two major percentiles below the previous values over a short period, (e.g. from 75th to 25th percentile).
While weight is the defining criteria for FTT, other anthropometric parameters, e.g. length/height for age and body mass index, are also frequently affected and may be used for diagnosis.
Etiologically: FTT may be due to non-organic, organic or mixed causes (Table 3.1).
TABLE 3.1: Causes of failure to thrive
Non-organic
a. Emotional deprivation
• Maternal death/chronic illness/separation
• Family disharmony
• Unwanted child
b. Nutritional: Protein-energy malnutrition
Organic
a. Infections: TB, chronic malaria, kala-azar, AIDS
b. GIT: Cleft palate, GER, malabsorption syndrome
c. Hepatic: Chronic liver disease, cystic fibrosis
d. Resp: Recurrent infections, asthma, chronic lung disease
e. CVS: CCF, cyanotic CHDs, infective endocarditis
f. CNS: Cerebral palsy, mental retardation
g. Renal: Chronic UTI, renal failure, renal tubular acidosis
h. Endocrinal: GH deficiency, cong. adrenal hyperplasia
i. Collagen disorders: Rheumatoid arthritis, SLE
j. Hematological: Severe anemia, malignancy
GER: Gastroesophageal reflux; GH: Growth hormone
a. Non-organic FTT is more common (80%), usually seen in under-five children due to dietary deficiencies or emotional deprivation.
b. Organic FTT may develop at any age due to identifiable causes, e.g.
- Decreased food intake due to difficulties in sucking, eating or swallowing.
- Impaired digestion/absorption of nutrients, e.g. malabsorption syndromes.
- Increased metabolic requirements, e.g. chronic diseases or infections.
- Increased losses of ingested food, e.g. chronic diarrhea or vomiting.
Clinically these children present with:
• Malnutrition, e.g. growth failure, anemia, vitamin and mineral deficiencies,
• Behavioral changes, e.g. apathy, social withdrawal, poor eye contact/response to cuddling,
• Developmental retardation,
• Recurrent or persistent infections,
• Signs of primary disease, e.g. chronic infections, illnesses, emotional deprivation, etc.
Diagnostic assessment of a child with FTT may be divided into three steps: (a) preliminary clinical evaluation and investigations, (b) evaluation of response to the trial feeding and (c) revaluation with more exhaustive investigations in cases, refractory to trial feeding (Fig. 3.1).
Step I. Preliminary evaluation includes:
a. Detailed history, specially related to dietary intake, preceding infections, e.g. diarrhea, child abuse/ neglect and developmental history. Available case records, e.g. growth charts should be reviewed to confirm the presence of FF and assess the age of onset.
b. Physical examination, specially related to anthropometric values and signs of malnutrition, vitamin/mineral deficiencies, systemic infections/illnesses, etc.
c. Developmental and psychosocial assessment to search for the cause, e.g. emotional deprivation as well as consequence of FTT.
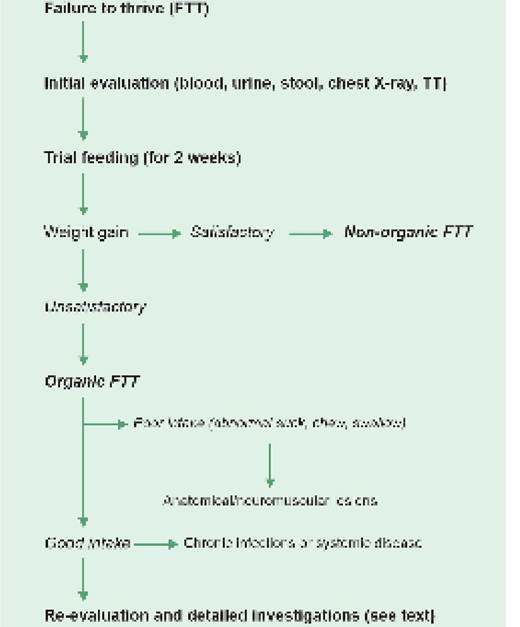
Fig. 3.1: Diagnostic approach in failure to thrive.
d. Baseline investigations to exclude common causes, i.e.
- Complete hemogram for anemia, infections, etc.
- Urine analysis for UTI, chronic renal disease, etc.
- Stool analysis for malabsorption, worms, etc.
- X-ray chest and tuberculin test for tuberculosis.
- Skeletal survey to assess bone age.
Step II. Trial feeding: While most cases may be managed at home with nutritional counseling and periodic followup, hospitalization is indicated in cases with—(a) severe undernutrition with weight and (d) doubtful dietary intake.
In hospital, these cases should receive trial feeding, i.e. supervised, unlimited high caloric diet (150-200 cal/kg/ day) for minimum 14 days, if necessary by nasogastric tube, along with daily weight record. A weight gain of ~50 g/day from 4-5th day onward and sustained for at least a week is considered as satisfactory, suggestive of non-organic etiology. Absence of satisfactory weight gain on trial feeding indicates organic FTT.
Step III. Re-evaluation with detailed investigations is indicated in non-responsive cases to trial feeding and includes:
• Biochemical investigations, e.g. blood sugar, serum proteins, liver/ renal function tests, screening tests for renal tubular acidosis/aminoaciduria.
• Endocrinal studies, e.g. thyroid function tests, growth hormone assays including somatomedin C levels, cortisol levels, etc.
• Genetic studies for inborn errors of metabolism, including enzyme assays in selected cases.
In addition, all cases of FTT must.
Management of FTT aims not only to nutritional rehabilitation but also to resumption of appropriate emotional environment and treatment of the underlying organic cause. A multi-disciplinary approach is necessary in most cases, including:
a. Nutritional management to correct the dietary deficit and allow catch-up growth, with: (a) increasing volume, frequency and caloric density of meals, (b) avoidance of low-caloric foods and (c) dietary and micronutrient supplementation;
b. Psychological support and modification of home environment;
c. Treatment of underlying cause and associated problems,
e. g. vitamin deficiencies, anemia, etc. All children should be immunized to their age-appropriate level;
d.
Parental counseling regarding: (a) correct nutritional practices including dietary requirements, (b) use of local nutritious foods, (c) correct cooking practices, (d) maintenance of dietary hygiene, (e) inculcation of good food habits in their children, (f) myths and misconceptions about foods, and (g) Psychosocial and emotional support to the child. They should be actively involved in nutritional rehabilitation process to ensure compliance with treatment and follow-up.e. Periodic growth monitoring and regular follow-up after discharge, as FTT frequently recurs due to continuance of etiological factors.
Prognosis: While initial catch-up growth is excellent in most of the adequately treated cases, it tends to slowdown over time and recurrence of FTT is not uncommon. Long-standing FTT in early life may lead to persistence of development problems, e.g. cognitive, behavioral and language disorders.
3.2 OBESITY
Body fat content changes from high adiposity state in infancy to the lowest level at 5-6 years of age, followed by gradual increase till adolescence. Obesity is a complex multi-factorial disorder characterized by an excess of adipose tissue, adversely affecting the health. While frequently used interchangeably, the terms overweight and obesity have different connotations.
Definition: Obesity and overweight in children > 2 years is defined on the basis of body mass index (BMI) as follows:
• Obesity: BMI above +3 Z-score (adult equivalent >27 kg/m2),
• Overweight: BMI above +2 to +3 Z-score (adult equivalent >23-27 kg/m2),
• Extremely obesity: 120% of the 95th percentile (adult equivalent ≥35 kg/m2).
In younger children < 2 years of age, obesity is considered when the weight for length exceeds 97th percentile.
Prevalence: Recent years have witnessed sharp rise in prevalence of obesity among children, reported in ~5% of those above 5 years of age, increasing with age.
Etiology: Obesity results from both hereditary as well as environmental factors.
It may be either exogenous due to life-style factors (95%) or endogenous due to pathological causes (5%) (Table 3.2). Pathological obesity is usually central in distribution, associated with dysmorphism, growth failure and delayed bone age.Exogenous (constitutional) obesity denotes an imbalance in dietary intake (high-fat, high-sugar, high-salt, energy- dense diet) and physical activity (sedentary life-style). Recent trends of excessive consumption of energy-dense junc foods, high screen-time and limited outdoor play have fuelled pandemic of obesity in children.
Some children are inherent prone for constitutional obesity due to genetic factors (Polygenic obesity) associated with single nucleotide polymorphisms of PSMA6 and PSMA3 proteosomal genes.
Endogenous (Secondary) obesity is due to pathological causes, e.g. endocrinal disorders, hypothalamic lesions and syndromic defects, e.g. Prader-Willi syndrome. Monogenic obesity due to loss of function mutations in leptin (LEP) and leptin receptor (LEPR) genes is more
TABLE 3.2: Causes of obesity in children
A. Exogenous (95%)
• Physiological in early adolescence
• Constitutional or familial
• Over-eating (behavioral)
• Poor physical activity:
♦ Habitual
♦ Pathological: Physical handicaps
B. Endogenous (1.0 in males and > 0.85 in females suggesting obesity.
Assessment of a child with obesity aims to identify the cause as well presence of complications and comorbidities, to plan appropriate management. It includes:
a. Detailed history to assess:
± Dietary intake by 24-hour dietary recall, with special reference to consumption of junk foods, binge-eating, frequent-snacking, etc.
± Physical activity in preceding few days including screen time;
± Presence of family obesity, life-style and motivation;
± Other risk factors, e.g. maternal gestation diabetes, early infant feeding, drug consumption, delayed development, etc.
b. Physical examination to:
± Confirm the presence of obesity, using anthropometric assessment and BMI charts;
± Detect markers of underlying diseases, e.g.
endocrinal or genetic disorders,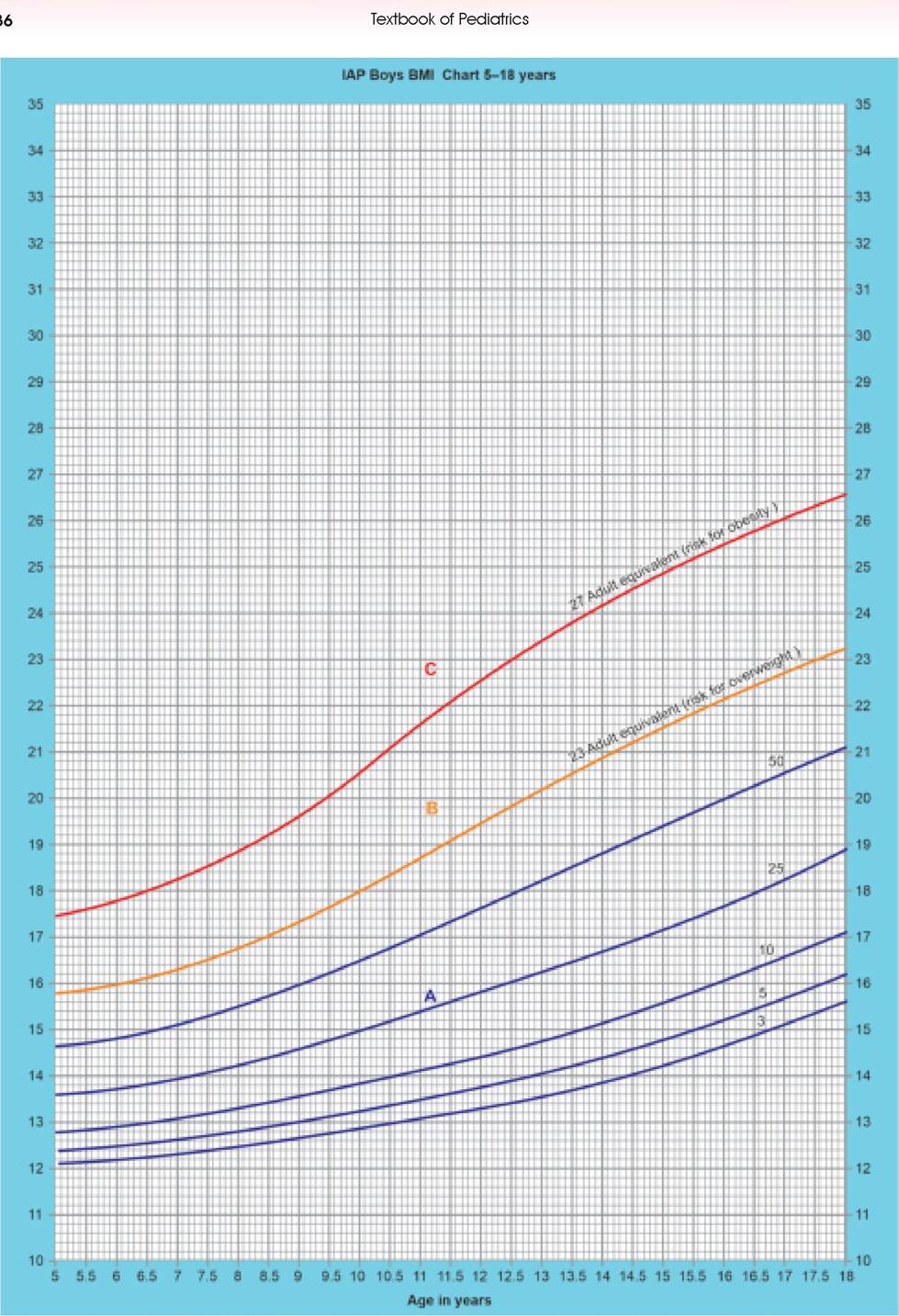
Fig. 3.2: Interpretation of child's BMI on BMI chart. (A) Normal; (B) Overweight; (C) Obesity
- Search for complications, e.g. sleep problems, hypertension, metabolic problems, etc.
c. Psychosocial assessment for body-image, self-esteem and secondary behavioral problems.
d. Relevant investigations to search for the cause of obesity as well as metabolic complications, e.g. glucose tolerance test, lipid profile, hormonal profile, etc.
Management of obesity in children should aim for gradual and sustained control with weight loss not exceeding 1.5 kg per month or >7-10% over 6 months, with a multidisciplinary approach involving pediatrician, dietician and behavioral counselor, as follows:
• Dietary modification, with low-caloric, low-fat, normal-protein, high-fiber diet, tailored to reduce the weight by ~500 g/week, is enough in most cases of overweight or mild obesity. Severe dietary restrictions and aggressive weight reduction plans are undesirable in children and may hamper normal growth. Junk foods and carbonated drinks should be avoided.
• Encouragement of physical activity, e.g. outdoor games or swimming, is more acceptable to children than rigid exercise schedules.
• Treatment of identifiable cause.
• Psychosocial support to the child and family.
• Drugs: General anti-obesity drugs, e.g. orlistat (gastric lipase inhibitor) or sibutramine (neurotransmitter modulator) affect growth and should be avoided in children. However, leptin (in leptin deficiency), octeotride (in hypothalamic obesity) and metformin (in polycystic ovarian syndrome) may be considered in selected cases.
• Surgery like gastric banding or jejuno-ileal bypass is rarely indicated except in morbid obesity with BMI >40 kg/m2.
• Referral: Children with-(a) early-onset obesity < 5 years age, (b) rapid progression, (c) delayed development, growth, and puberty, (d) neurological features and (e) abnormal metabolic workup should be referred for detailed evaluation and management under expert guidance.
Prevention: Obesity in adults usually originates in childhood and prevention should begin as early as possible with appropriate guidance and monitoring. Important preventive interventions evention of obesity in childhood include -
a. Dietary modifications to restrict high-fat, high- sugar, high-salt diet; and encourage consumption of whole grains, legumes, fruits, vegetables and nuts. JUNCS food should be avoided as per IAP guidelines (Table 3.3). Fibre content in diet should be ~5 g + age in years. Exclusive breastfeeding till 6 months followed by appropriate complementary feeding is known to curtail risk of obesity in later life.
TABLE 3.3: IAP Guidelines on consumption of JUNCS* food
• Avoid JUNCS foods or limit to one serving/ week with
< 50% of total energy intake
• Limit dietary fat to 2 years (No restriction < 2 years)
• Eliminate trans-fat from diet
• Restrict free sugar content to evaluation and referral to specialized services is indicated in children with:
TABLE 3.4: Common causes of short stature
A. Normal variants short stature
• Familial
• Constitutional
B. Primary short stature (primordial dwarfism)
• Intrauterine growth retardation (IUGR)
• Genetic:
- Chromosomal: Mongolism, Turner syndrome
- Inborn errors of metabolism: Hurler syndrome
- Others: Progeria, Silver-Russell syndrome
• Skeletal dysplasia
- Short-limb: Achondroplasia
- Short-trunk: Spondylo-epiphyseal dysplasias
C. Secondary short stature
• Nutritional: PEM, zinc deficiency
• Emotional: Psychosocial dwarfism
• Chronic infections: TB, AIDS, UTI, worms
• Chronic systemic diseases:
- GIT: Malabsorption syndrome, chronic liver disease
- Renal: Chronic renal failure, renal tubular acidosis
- Cardiac: Cyanotic CHDs, recurrent CCF
- Pulmonary: Asthma, tuberculosis
- CNS: Diencephalic syndrome of infancy
- Blood: Hemoglobinopathies, malignancies
• Endocrinal disorders:
- Growth hormone deficiency
- Hypothyroidism
- Cong. adrenal hyperplasia, Cushing syndrome
• Iatrogenic: Steroids, cytotoxics, radiotherapy
a. Severe short stature with height disease, Turner/ Noonan syndrome, intrauterine growth retardation and Idiopathic SS, etc. It should be started as soon as possible after confirmation of diagnosis, using a recombinant GH preparation as daily night injections (25-50 μg∕kg, subcutaneously). Higher doses may be used during puberty. Maximum response is seen during first year of therapy which must be continued till height velocity drops 14 years in girls and >16 years in boys.
Pseudotumor cerebri is a significant but rare complication of GH therapy. Rare side-effects include fluid retention, impaired glucose tolerance, worsening of scoliosis and slipped capital femoral epiphysis. Very rarely, there is increased risk of malignancy in some predisposed genetic conditions.
3.4 TALL STATURE
Tall stature, defined as height >97th percentile for the corresponding age is relatively uncommon.
Some children may appear taller than others in early childhood due to individual variations and ectomorphic body build. Hildren with precocious puberty are relatively taller in early puberty than others, though final adult height in them is normal due to early skeletal maturation and completion of puberty.
Etiology: Most of these cases are normal variants due to familial or constitutional features, though pathological tall stature indicates chromosomal, genetic or endocrinal etiology (Table 3.6). Some unique causes of tall stature, not discussed elsewhere, are as follows:
Marfan syndrome is the commonest cause of pathological tall stature in children. It is an autosomal dominant connective tissue disorder, with abnormal synthesis of fibrillin-1, an α-glycoprotein in elastin fibers.
TABLE 3.6: Causes of tall stature
• Familial (tall parents)
• Constitutional
• Genetic
- Chromosomal: Klinefelter or XXY syndrome
- Genetic: Marfan syndrome, homocystinuria
• Endocrinal
- Hyperpituitarism (pituitary gigantism)
- Precocious puberty
- Others: Thyrotoxicosis, Beckwith syndrome
• Cerebral gigantism (Soto's syndrome)
Clinically, these cases are characterized by tall stature, increased arm span (US:LS ratio), arachnodactyly (long fingers), ocular abnormalities, (e.g. lens dislocation, iridodonesis) and cardiac abnormalities, (e.g. mitral valve prolapse, aortic regurgitation).
Cerebral gigantism (Soto's syndrome) is a rare hypothalamic disorder, characterized by rapid linear growth during first 3-4 years of age, macrocrania, mental retardation, gait abnormalities and normal or precocious puberty. It may be differentiated from pituitary gigantism by normal growth hormone levels and delayed skeletal maturation as compared to height.
Diagnostic evaluation of tall stature aims to differentiate normal variants form rare pathological causes, usually based on positive family history (correlate with mid- parental height) and normal physical examination. A suspected cases of pathological tall stature needs complete clinical evaluation, including ophthalmic and cardiac assessment (for Marfan syndrome and homocystinuria) and detailed investigations, including:
a. Radiological determination of bone age.
b. Karyotyping to exclude chromosomal disorders, e.g. Klinefelter syndrome.
c. Hormonal studies, e.g. growth hormone assays and thyroid function tests.
Treatment: No treatment is usually necessary for tall stature per se, except the treatment of underlying cause. Hormonal therapy with sex-steroids to accelerate epiphyseal fusion and early completion of puberty is occasionally used in children with severe tall stature (with predicted adult height >3 SD) and significant psychological stress.
3.5