HEMOPHILIA
Hemophilia is an X-linked recessive deficiency of either fVIII (classic hemophilia or Hemophilia A) or fIX (hemophilia B or Christmas disease)-the former being more common (80-85%).
Hemophilia A occurs in ~1#8725;5000 live male births.Both defects share common mode of inheritance as well as clinical presentation, but with differences in diagnosis and management.
Etiopathogenesis: fVIII and fIX are essential components of intrinsic clotting pathway, involved in activation of fX and absence of either of these proteins impairs the ability to generate thrombin and fibrin leading to delayed and friable clot formation.
Severity of these disorders depends on the percentage of normal factor activity, graded as: (a) mild haemophilia (gt;5%), (b) moderate hemophilia (1-5%), and (c) Severe hemophilia (lt;1%). One unit of each factor denotes the activity of the corresponding factor in 1 ml of normal plasma. Deficiency of von Willebrand factor can also lead to secondary deficiency of fVIII.
Clinical features: Being X-linked diseases, only males are affected, while females are carriers. While some cases may present at neonatal period with bleeding from umbilical stump or during circumcision (as fVII and fIX do not cross placenta), most cases manifest in infancy with:
• Recurrent spontaneous hemarthrosis (or after trivial trauma), usually at ankles, knees or elbow with pain and swelling is the commonest manifestation, leading to chronic deformities in older children (Fig. 19.11).
• Mucocutaneous bleeding, e.g. easy bruising, excessive and prolonged bleeding after trivial trauma, gum bleeds, intramuscular hematoma, etc.
• Life-threatening bleeding in severe disease from GIT, upper airways and intracranial space.
• Mild hemophilia may not develop spontaneous hemorrhages but bleed longer following trauma or surgeries and remain undiagnosed for many years.
Female carriers are usually asymptomatic, except those who either have reduced factor levels due to lyonization of X-chromosome or have only one sex chromosome, e.g. Turner syndrome (XO).
Laboratory diagnosis is based on:
• Clotting time (CT) is prolonged in severe hemophilia, though normal CT does not exclude mild disease.
• Activated partial thromboplastin time (aPTT) is the most reliable screening test, which is usually three times higher than in control but can be corrected by mixing with normal plasma.
• Prothrombin time, an indicator of extrinsic pathway abnormality, is normal in hemophilia as also the BT, TT and platelet count.
• Specific factor assays, though not essential, are useful for diagnostic confirmation and quantification of fVIII and fIX activity.
TABLE 19.24: D/D of coagulation disorders
Inherited clotting factor deficiencies
• Hemophilia A (fVIII)
• Hemophilia B (fIX)
• Hemophilia C (fXI)
• Parahemophilia (fV)
• von Willebrand disease
• Others: (fVII, X, prothrombin, XIII, fibrinogen)
Acquired (vitamin K dependent) factors* deficiencies
• Hemorrhagic disease of newborn
• Chronic liver disease/malabsorption
• Prolonged antibiotic therapy
• Disseminated intravascular coagulation (DIC)
• Anti-coagulant therapy
*fII, VII, IX, X
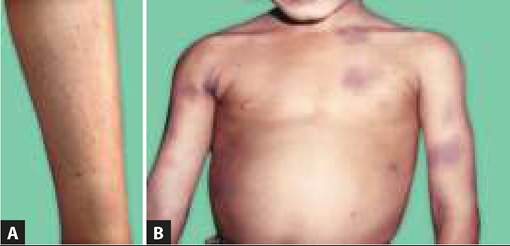
Fig. 19.11: Hemophilia: (A) Mucosal bleeding; (B) Hemoarthrosis.
fVIII has two components — antigenic component (fVIII-Ag) and coagulant component (fVIII-C). In hemophilic cases or carriers, fVIII-C is reduced but fVIII-Ag is normal (d/d von Willibrand disease, in which fVIII-Ag activity is reduced).
• Ancillary investigations, e.g. joint X-rays and neuroimaging for intracranial bleeds may be required in cases with chronic arthropathy or CNS signs.
Antenatal diagnosis of hemophilia is possible at ~18- 20 weeks by assessment of fVIII#8725;fIX activity in cord blood sample.
However, many cases could be identified earlier at 14-16 weeks by molecular studies in amniotic fluid; or even at 8-10 weeks by chorionic villous sampling.Carrier detection in Hemophilia A is based on fVIII-C and fVIII-Ag ratio. A ratio of lt;0.6 indicates carrier state in females. Molecular diagnosis can also be used identify genetic mutation in carriers.
Management: Comprehensive management of hemophilia is a team-work involving physician, nurse, orthopedic surgeon, physiotherapist and psychosocial workers, which aims to control acute bleeding episodes and minimize long-term joint disabilities to improve the quality of life. Important steps in management include:
I. Control of acute hemorrhagic episodes revolves around the factor replacement therapy though local pressure/ packing and anti-fibrinolytic therapy (discussed below) may be helpful in minor bleeds.
• Factor replacement therapy aims to increase corresponding factor levels above the hemostatic level, i.e. 35-40% (for fVIII) or 25-30% (for fIX), in mild/ moderate bleeds and up to 100% in life-threatening intracranial bleeding. However, the options for factor replacement differ in fVIII and fIX deficiency.
- fVIII deficiency is best corrected by purified fVIII concentrates, which may be plasma-derived or recombinant. Recombinant products are preferred due to less risk of transfusion-related infections. Each bag of fVIII concentrate contains ~200 units and the desired dose is calculated as follows:
fVIII (Units) = (desired fVIII%) ? Body weight (kg) ? 0.5 For example, to achieve plasma level of 40% in a 15-kg child, calculated dose is 40 15 0.5 = 300 units. Each unit of fVIII#8725;kg increases the level by 2% with half-life of 8-12 hours.
Cryoprecipitate may also be used for fVIII replacement, if concentrates are not available. Each bag of cryoprecipitate contains ~100 units of fVIII but carries the potential risk of volume overload, transfusion-related infections or allergic reactions due to presence of human protein.
- fIX deficiency may be corrected by purified fIX concentrates (calculated as above), prothrombin complex concentrates or fresh frozen plasma.
Cryoprecipitate is of no use due to little fIX activity.Considering short half-life of fVII and fIX, these factors need to be transfused every 8-12 hours to sustain desired levels,. However in hemarthrosis and closed-space muscle bleeds, bleeding usually stops with one or two transfusions and does not recur due to surrounding pressure. Treatment of various types of bleeds in hemophilia is shown in Table 19.25.
Development offVIII or #8739;X antibodies (inhibitors) should be suspected in cases, which do not respond to adequate factor replacement. These antibodies usually develop after repeated transfusions and estimated by quantitative assays (in Bethesda units).
TABLE 19.25: Factor replacement therapy for bleeding episodes in hemophilia
1Till recovery or return to baseline joint functions, 2Total duration of therapy
*fIX doses for plasma-derived products, 30% higher for recombinant products
These patients may respond to higher doses of fVIII orf IX or need to be treated with activated prothrombin complex concentrates or aPCC (fVIII inhibitor bypassing activity or FEIBA) or recombinant fVIIa (Novoseven).
A new therapeutic option for hemophilia A with/without inhibitors includes Emicizumab, a monoclonal antibody, which performs the function of fVIII by bridging fIXa and fX, mimicking activated fVIIIa.
• Supporting measures to control active bleeding in hemophilia include:
± Non-factor substitutes: Desmopressin acetate (DDAVP) releases endogenous fVIII and may be used during elective dental/minor surgical procedures in cases of mild/moderate hemophilia A or von Willebrand disease, but not in severe hemophilia A or B. It is given as 0.3 #956;g#8725;kg IV in 2550 ml normal saline over 20-30 minutes or as nasal spray. A fibrin glue has been used to seal bleeding sites in minor procedures, e.g.
dental extraction.± Anti-fibrinolytic therapy is used to control mucosal bleeds or for dental extraction, using Tranexemic acid (PO 25 mg/kg 6-hrly) or Epsilon aminocaproic acid or EACA (PO 100-200 mg/kg initially followed by 50-100 mg/kg 6-hrly)
± Local measures, e.g. pressure packing/bandage or cold compresses, etc. may be helpful in cases with superficial mucosal/skin bleeds.
II. General care in these cases include:
• Prevention of bleeding by avoidance of physical trauma, e.g. contact-sports, IM injections, etc., drugs, e.g. aspirin and good dental care.
• Correction of anemia, if necessary, by blood/ packed cell transfusions.
• Prevention of transfusion related infections by proper screening of blood products, HBV vaccination, and periodical HIV/HBV screening.
• Symptomatic therapy with bed-rest, joint immobilization, cold-compresses and codeine during acute hemarthroses. Analgesics, e.g. aspirin, may inhibit platelet functions and should be avoided. Joint aspirations should be avoided except in severe persistent effusions, and should be preceded by preaspirate factor infusion.
• Prevention of joint deformities by physiotherapy between attacks and surgical corrections.
• Social and psychological support.
• Rehabilitation and occupational therapy.
• fVIII prophylaxis (15-20 IU/kg thrice a week) is also used in severe haemophilia to prevent deformities, frequent needs of hospitalizations and improve the quality of life.
III. Genetic counseling with carrier detection and prenatal diagnosis, using molecular studies on chorionic villus biopsy sample or amniocentesis.
Long-term complications of hemophilia include: (a) chronic arthropathy, deformities and disabilities, (b) transfusion-related complications and infections, (c) psychological disorders, and (d) development of fVIII or fIX antibodies.
Other important but milder coagulation disorders include:
von Willebrand disease (vWD) is the commonest hereditary bleeding disorder, accounting for ~3-5% of all coagulation defects.
Etiology: vWD is an autosomal dominant disorder involving chromosome 12, characterized by deficiency of von Willebrand factor (vWf) — a carrier protein for fVIII, which also has a role in platelet aggregation.
Consequently, these cases are characterized by combination of two hemostatic defects—(a) fVIII deficiency and (b) platelet aggregation defect. Severity-wise, it is classified as Type I (vWf reduced), Type II (vWf function defect) and Type III (vWf absent).
Clinically most cases present with mild to moderate mucocutaneous bleeding beyond neonatal period. Some cases present only as menorrhagia in adolescents. However, type III may present with severe bleeding in newborns or spontaneous hemoarthrosis in later life.
Diagnosis is supported by a combination of prolonged bleeding time (platelet defect) and PTT (clotting factor defect), confirmed by quantitative vWf assay. Unlike Hemophilia A, fVIII-Ag activity on Ristocetin assay is reduced in vWD.
Treatment: Mild cases may be treated with antifibrinolytic agents, e.g. EACA or non-factor substitutes, e.g. DDAVP, while severe cases need factor replacement with intermediate-purity fVIII concentrates (which also contain vWf) or cryoprecipitate.
Hemophilia C (factor XI deficiency) is an autosomal recessive disorder with mild to moderate bleeding tendency. Replacement therapy is usually required only during surgical procedures, using fresh frozen plasma, as this factor is not available in cryoprecipitate or purified concentrate form.
19.13
More on the topic HEMOPHILIA:
- 4 Preconception Counseling and Prenatal Care
- Conclusion
- Macrovascular Complications of Diabetes Mellitus
- Etiopathogenesis
- Chapter 7 Genetics and Genetic Disorders in Obstetrics and Gynecology
- REFERENCES
- PRECONCEPTION AND interconception care ^292
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025