Physical Examination
Physical examination findings help focus further diagnostic evaluation, utilizing such tools as electrodiagnosis, molecular genetic testing and histopathologic analysis of biopsy specimens.
All diagnostic information must be interpreted within the context of relevant clinical information. In many instances, a precise molecular genetic diagnosis is not medically possible. However, the accurate characterization of an individual patient within the most appropriate NMD clinical syndrome still allows the clinician to provide the patient and family with accurate prognostic information and anticipatory guidance for the future.Specific aspects of the physical examination, relevant to the neuromuscular disease population, includes simple inspection for the presence of focal or diffuse muscle wasting or focal enlargement of muscles, as with the “pseudohypertrophy” seen in such dystrophic myopathies as Duchenne and Becker muscular dystrophy (Fig. 12.1). The increase in calf circumference in DMD is caused by an increase in fat and connective tissue rather than true muscle fiber hypertrophy in the gastrocnemius. Over time, the reduced bulk of musculature may be caused by more severe fiber loss in a more “active” dystrophic process affecting proximal musculature. Other neuromuscular disorders may show calf pseudohypertrophy, such as childhood-type acid maltase deficiency.
Focal atrophy of particular muscle groups may provide diagnostic clues to specific neuromuscular disorders, such as spinal muscular atrophy, Emery-Dreifuss muscular dystrophy, FSHD, and LGMD. Those with CMT, particularly those with type II axonal forms, demonstrate distal atrophy or “stork leg appearance” relatively early in the disease course. Palpable nerves in the cubital tunnel, posterior auricular region, or around the fibular head may be indicative of “onion bulbs” seen in hereditary demyelinating neuropathies such as CMT I subtypes or Dejerine-Sottas disease (CMT III).
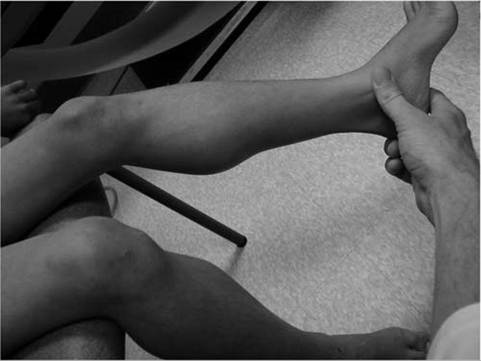
Figure 12.1 Calf pseudohypertrophy in a male with Duchenne muscular dystrophy.
Muscle fasciculations may be seen as nonspecific findings of a variety of lower motor neuron disorders. Fasciculations are particularly common in such lower motor neuron disorders as SMA. Distal fine tremor may be seen in a large proportion of CMT patients (30-50%), and in other patients with weakness such as SMA. “Polyminimyoclonus,” another variant of muscle fasciculations characterized by a fine tremor of the fingers and hands, may be evident in SMA I and II.
Infants with NMD often show infantile hypotonia (Fig. 12.2), the differential for which is large (see chapter on pediatric electrodiagnosis).
A thorough general physical examination of cardiac, pulmonary, and gastrointestinal systems should be performed on all patients suspected of having a neuromuscular disease. Hepatomegaly may be seen in such metabolic myopathies as acid maltase deficiency (type II glycogenosis or “Pompe disease”) and type III and IV glycogenosis. The skin should be evaluated
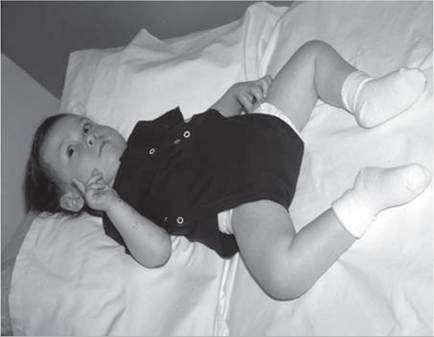
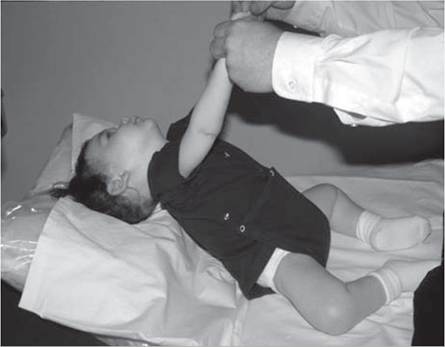
Figure 12.2 (A, B) Hypotonia in an 18-month-old child with spinal muscular atrophy.
for characteristic skin rashes and nail bed capillary changes if an inflammatory myopathy such as dermatomyositis is suspected. Keratosis pilaris is a characteristic skin rash seen in congenital muscular dystrophy with collagen VI deficiency (Fig. 12.3). Craniofacial changes and dental malocclusion are common in congenital myotonic muscular dystrophy, congenital myopathies, congenital muscular dystrophy, and type II SMA. A neurological examination should include a thorough evaluation of cranial nerve function, muscle tone, muscle strength, sensory and cerebellar function, and deep tendon reflexes.
An assessment for the presence of percussion and grip myotonia (Fig. 12.4) should be performed in situations where a myotonic syndrome is suspected. Musculoskeletal examination will reveal the presence of limb contractures, deformities, and spinal deformity.Some neuromuscular disorders, such as congenital myotonic muscular dystrophy (congenital DM1), Fukuyama congenital muscular dystrophy, selected cases with mitochondrial encephalomyopathies, and a small proportion of Duchenne muscular dystrophy cases, may have significant intellectual impairment.
A thorough functional examination is essential in the diagnostic evaluation of a patient with suspected neuromuscular disease. This includes an evaluation of head control, bed/mat mobility, transitions from supine-to-sit and sit-to-stand, sitting ability without hand support, standing balance, gait, stair climbing, and overhead reach.
An evaluation of overhead reach is performed while examining the patient from the front and from behind in order to evaluate shoulder girdle weakness. Careful assessment of scapular winging, scapular stabilization, and scapular rotation is helpful in the assessment of patients with FSHD or other limb girdle syndrome. The scapula is stabilized for overhead abduction by the trapezius, rhomboids, and serratus anterior. Abduction to 180 degrees requires strong supraspinatus and deltoid muscles in addition to strong scapular stabilizers.
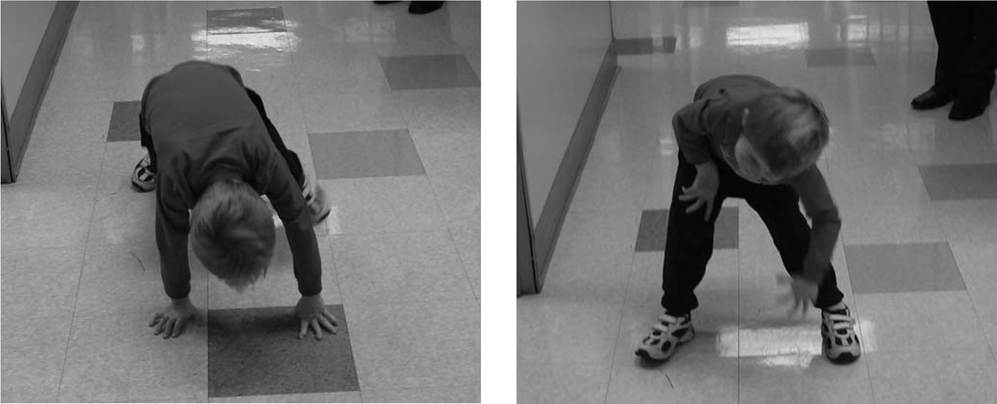
Patients with proximal weakness involving the pelvic girdle muscles may rise off the floor using the classic “Gower's sign,” where the patient usually assumes a fou- point stance on knees and hands, brings the knees into extension while leaning forward the upper extremities, substitutes for hip extension weakness by pushing off the knees with the upper extremities, and sequentially moves the upper extremities up the thigh until an upright stance with full hip extension is achieved (Fig. 12.5). A Gower's sign is not specific to any neuromuscular condition, but may be seen in a variety of neuromuscular diseases, including DMD, LGMD, SMA type III, severe childhood autosomal recessive muscular dystrophy (LGMD II subtypes), congenital muscular dystrophy, congenital myopathy,
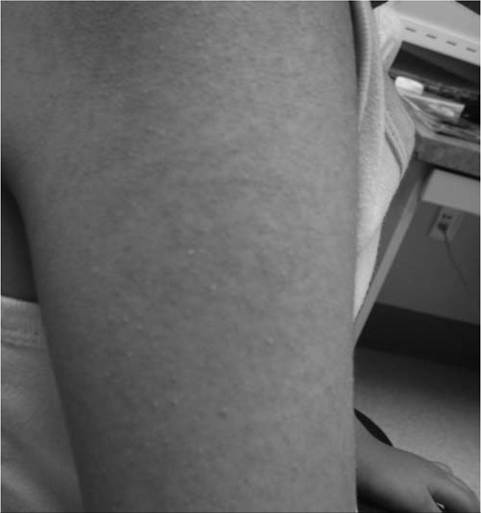
Figure 12.3 Keratosis pilaris skin rash on the extensor surface of the arm in Ullrich congenital muscular dystrophy with collagen type VI abnormality.
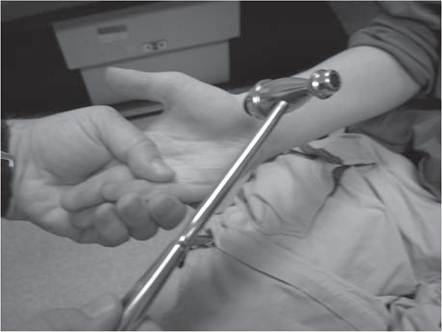
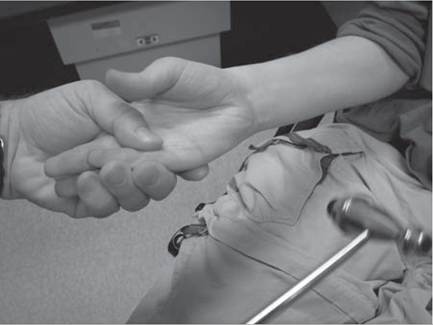
Figure 12.4 (A-C) Percussion myotonia of the thenar eminence.
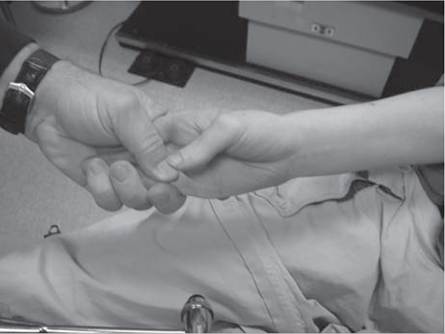
myasthenic syndromes, severe forms of CMT (eg, CMT III and CMT IV), and in other neuromuscular disease conditions producing proximal weakness. Patients with proximal lower extremity weakness often exhibit a classic myopathic gait pattern (Fig. 12.6). Initially, weakness of the hip extensors produces anterior pelvic tilt and a tendency for the trunk to be positioned anterior to the hip joint. Patients compensate for this by maintaining lumbar lordosis, which positions their center of gravity/weight line posterior to the hip joints, thus stabilizing the hip in extension on the anterior capsule of the hip joint. Subsequently, weakness of the knee extensors produces a tendency for patients to experience knee instability and knee buckling
with falls. Patients compensate for this by decreasing stance-phase knee flexion, and they posture the ankle increasingly over time into plantar flexion. This produces a knee extension moment at foot contact and plantarflexion of the ankle during mid- to late-stance phase of gait, which helps position the weight line/ center of gravity anterior to the knee joint (thus producing a stabilizing knee extension moment). Patients with DMD will progressively demonstrate toe walking with initial floor contact, with the foot contact increasingly moving forward onto the midfoot and finally the forefoot as they reach the transitional phase of ambulation before wheelchair reliance (see Fig. 12.7). Finally, weakness of the hip abductors produces a tendency toward lateral pelvic tilt and pelvic drop of the swing phase side. Patients with proximal weakness compensate for this by bending or lurching the trunk laterally over the stance-phase hip joint (Fig.
12.8). This produces the so-called “gluteus medius lurch” or Trendelenburg’s gait pattern.Patients with distal weakness affecting the ankle dorsiflexors and ankle everters and less severe proximal

A

B
C D
Figure 12.5 (A-F) Gower's sign in a seven-year-old boy with Duchenne muscular dystrophy due to hip extension weakness.

E
Figure 12.5 Continued
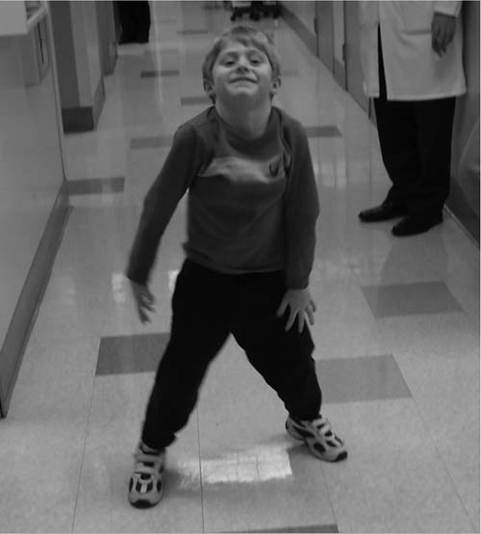
F
weakness (eg, CMT, Emery-Dreifuss muscular dystrophy, myotonic muscular dystrophy, FSHD, and other conditions) often exhibit a foot slap at floor contact with a steppage gait pattern to facilitate swing-phase clearance of the plantar-flexed ankle. Alternatively, these patients may clear the plantar-flexed ankle using some degree of circumduction at the hip or vaulting on the stance-phase side. Milder distal lower extremity weakness may become clinically evident by testing heel walking and toe walking.