CHAPTER 43 TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES
DOLORES GAVIER-WIDEN
National Veterinary Institute (SVA), and Swedish University of Agricultural Sciences, Uppsala, Sweden
Transmissible spongiform encephalopathies (TSE), or prion diseases, are a group of infectious neurodegenerative diseases caused by a transformed host protein.
These transformed proteins are called prions (proteinaceous infectious particles), a novel type of infectious agent that is devoid of nucleic acid.I n animals TSE include bovine spongiform encephalopathy (BSE), atypical BSE, scrapie, atypical scrapie or Nor98, chronic wasting disease (CWD), transmissible mink encephalopathy (TME), feline spongiform encephalopathy (FSE); in humans TSE include, among others, Creutzfeldt-Jakob disease, with the sporadic (sCJD), variant (vCJD), iatrogenic and familial subtypes.
AETIOLOGY
The cellular prion protein (PrPc) is a glycosylphosphatidylinositol-linked copper-binding membrane protein of approximately 220 amino acid residues typically in α-helical conformation. The cellular prion protein is a normal cell surface protein of neurons and other cells encoded by a unique Prnp gene; it is highly conserved across diverse species. The disease-causing isoform of PrPC is named PrP scrapie (PrPSc), and the terms PrPcwD and PrPBSE are used to denote the source. This abnormal relatively protease-resistant isoform of the PrP protein is rich in β -pleated sheets. PrPc is converted into PrPSc during a process of conformational change, which results in replacement of some of the α-helical structure by β-sheets. This form can adopt a fibrillar aggregated structure, which is a feature of many of the deposits observed in the brain of animals with prion diseases. This post-translational alteration of PrPc into the pathogenic infectious isoform, PrPSc, is considered to be the principal molecular basis of TSE.
The ‘protein only’ hypothesis proposed that PrPSc was the cause of prion diseases, that it was infectious on its own and that it replicated by interacting with the cellular prion protein of the host so that PrPc would undergo a conformational conversion into PrPSc(1). This proteinase K (PK)-resistant PrPSc accumulated in the tissues, as it could not be degraded as the normal cellular prion protein(1) and became the most effective marker of prion infection. The fraction resulting from the treatment of PrPSc with PK is termed PrPres (‘res’ meaning resistant) and most diagnostic methods are based on its detection. PrPSc cannot be inactivated with formalin, is highly resistant to traditional sterilization methods such as autoclaving and disinfecting and it has an extraordinary environmental persistence. The BSE agent, like all the human TSE agents, is categorized as a Hazard Group 3 pathogen(2), but as there is no evidence of airborne transmission, full containment Level 3 is not required.
Infectious Diseases of Wild Mammals and Birds in Europe, First Edition. Edited by Dolores Gavier-Widen, J. Paul Duff, and Anna Meredith. © 2012 Blackwell Publishing Ltd. Published 2012 by Blackwell Publishing Ltd.
Biochemically, the properties of PrPSc appear to be determined by the three-dimensional shape of the molecule, changes to which could result in different forms, or ‘strains’.
EPIDEMIOLOGY
GEOGRAPHICAL DISTRIBUTION AND HOSTS
The first naturally occurring TSE to be recognized was scrapie, with descriptions dating back to the 18th century. The susceptibility of sheep to scrapie has been shown to be heavily dependent on the Prnp genotype of the host. Programmes for selective breeding for scrapie resistance have been implemented in many countries as part of a control strategy and current EU control policies advocate culling on a selective genotype basis.
Nor98, or atypical scrapie, is a novel TSE type first recognized and described in Norway in 1998)3) and later identified as sporadic cases in most European countries as well as in the USA, Canada and New Zealand/4) It has been postulated to be a spontaneous form of TSE in animals. Nor98 affects old sheep and goats of more scrapie-resistant genotypes but not of the most susceptible genotypes. No cases of scrapie or atypical scrapie have been reported in wild Caprinae or Ovidae in any continent.BSE was first identified in the UK in 1986(5) and was propagated within cattle by recycling meat and bone meal from infected cattle. This resulted in a large-scale epidemic in the mid 1990s, which has to date affected approximately 184,600 cattle in the UK and more than 7,900 cases outside the UK, including 13 European member countries, Japan, Canada and the USA. In 1996, vCJD in humans was shown to be caused by BSE, probably by consumption of contaminated beef products. In the EU, the BSE concern led to the introduction of a series of measures aimed at its eradication, including large- scale testing in EU member states.
BSE is unique among the TSE in that it has extensively crossed the species barrier. Besides the human form, vCJD, natural BSE infection has occurred in seven species from the family Bovidae, four Felidae and four non-human primates in zoological collections)6-8). FSE also has been shown to be a BSE infection of domestic cats)9). Sheep and goats orally inoculated with BSE develop a TSE pathologically and clinically similar to scrapie)10). It is also well established that small ruminants in Europe received feed supplements containing meat and bone meal over the time period that cattle were exposed. This finding raised the concern that possibly cases diagnosed as scrapie could actually be BSE infection in sheep, and that endemic scrapie may mask BSE infections within the sheep population, and it questioned the safety of consuming sheep meat.
This resulted in obligatory large- scale active surveillance of small ruminants in the EU, implemented in 2002)11). Since 2005, all the TSE cases in small ruminants in the EU are required to undergo discriminatory testing to determine if the TSE is scrapie or BSE )11). Up until October 2011 only one TSE-positive case in a small ruminant, a domestic goat in France, has been confirmed to be BSE)12).Atypical BSE is a novel presentation of TSE in old cattle, with unusual molecular and pathological features. Two main types have been identified: BSE-L and BSE-H. ‘ L’ stands for low and ‘H’ for high, making reference to the differences in the molecular size of the PrPSc unglycosylated protein band seen on western blot )WB) assays compared with that of BSE. More than 60 cases of atypical BSE have been detected globally. It has been hypothesized that atypical BSE is a spontaneous form of TSE. No cases of BSE or atypical BSE have been reported in free-ranging wildlife.
TME was at first recognized as a food-borne disease of ranch-raised mink )Mustela vison) in the USA and was thereafter detected from time to time in several other countries, but it has not been detected in wild or farmed mink in Europe.
CHRONIC WASTING DISEASE
CWD was first recognized as a clinical entity of unknown cause in cervids in 1967 in captive mule deer )Odocoileus hemionus), derived from free-living populations in Colorado, USA)13). A similar syndrome was identified in Wyoming, USA, in 1978 and in the same year spongiform encephalopathy was found in affected animals from Colo- rado)13). Since its first recognition as a TSE, the geographical distribution and number ofcases ofCWD in free- ranging and captive deer herds within North America has increased significantly. As far as it is known, CWD is still restricted to North America, with the exception of a report in South Korea in captive elk imported from Canada. In North America, CWD ( Cervus elaphus nelsoni) occurs in 17 states in the USA and in the provinces of Saskatchewan and Alberta in Canada.
However, it is likely that if CWD occurs in countries or areas not known to be affected it will not be identified unless specific surveillance and testing for CWD is done.Hosts
Chronic wasting disease has been identified in the following cervid species in North America: mule deer, whitetailed deer (Odocoileus virginianus), Rocky Mountain elk (Cervus elaphus nelsoni) and Shira’s moose (Alces alces shirasi). CWD is not known to affect other species naturally, including carnivores and scavenger species. CWD has been transmitted to several other species, including cattle, by intracerebral inoculation, but oral experimental inoculation with CWD did not result in infection(14’15).
In Europe, three species from the family Cervidae have been considered as possibly susceptible to CWD owing to their taxonomic relation to the known target species. These are the European red deer ( Cervus elaphus elaphus), the European moose (Alces alces alces), and the white-tailed deer, which were introduced into Finland from North America in the 1930s.
Few experimental studies have been conducted to assess the susceptibility of European cervids to TSE. In a study in Canada, CWD was transmitted to red deer (Cervus elaphus elaphus) of the same subspecies as the European red deer by oral inoculation, indicating likely susceptibility of red deer in Europe(16). In the UK, red deer (Cervus elaphus elaphus) were inoculated with BSE orally or intracerebrally. This resulted in a TSE with different immunohistochemical and biochemical properties from CWD(17’18).
As in other TSEs, susceptibility to CWD also appears to be related to polymorphisms in the Prnp gene. A study on Prnp variability was conducted on 715 wild animals in Europe, including European red deer ( Cervus elaphus), roe deer (Capreolus capreolus) and chamois (Rupicapra rupic- apra). In the red deer, the study identified a number of polymorphisms giving rise to 12 haplotypes.
One of the haplotypes was identical to the Prnp of Rocky Mountain elk. In chamois one silent mutation at codon 119 was found. In roe deer, no single nucleotide polymorphisms were detected(19). The study concluded that the species investigated had a Prnp genetic background compatible with susceptibility to TSE.THE EUROPEAN CWD SURVEY
A survey for CWD and other TSE in farmed and wild cervids in the EU was set up by the European Commis- sion(20). This was the first time a European-wide survey for TSE was conducted in wild animals. Until then, little information was available on TSE diagnosis in cervids in Europe and the occurrence of CWD could not be known without specific testing. Also, it was known that farmed cervids in Europe had been fed compound feed (containing ruminant protein); therefore, the risk for BSE exposure could not be ruled out(21). The design of the survey was based on European Food Safety Authority (EFSA) opinion in 2004(21) which at that time indicated that in Europe red deer and white-tailed deer were the only two species considered most likely to be susceptible. North American moose were found to be susceptible to natural CWD first in 2005(22), and therefore the susceptibility of European moose was postulated only after the survey had been designed(20).
All EU member states were required to test as many farmed and free-living cervids as possible from all species and older than 18 months, of the following risk categories: cervids with signs of disease (such as neurological signs or poor body condition); cervids fallen (found dead) or culled and road-injured or road-killed cervids. Additionally, member states with large populations of farmed or free- living red deer, and Finland, the only European country with known white-tailed deer populations, needed to test a minimum number of healthy animals harvested by hunting.
All rapid tests approved at that time for BSE monitoring could be used for screening for CWD/TSE in European cervids and immunohistochemistry (IHC) or WB would be used for confirmation. Even though EFSA recommended testing of both brainstem and lymph node(21), the European Commission decided to conduct the survey testing brainstem samples only, probably as a result of practical and logistic reasons, as most member states had well-developed routines for brainstem sampling based on the experience of BSE and scrapie surveillance.
The survey was conducted largely during the years 2007, 2008 and 2009, with some samples collected during 2006 and 2010. On the whole, 14 member states and Norway tested 3,274 farmed cervids, including the following numbers and species: 2,781 red deer, 348 fallow deer (Dama dama), 44 reindeer (Rangifer tarandus tarandus), 3 sika deer (Cervus nippon), 4 moose and 94 cervids of unknown species; and 21 member states and Norway tested 10,049 wild cervids, including 7,329 red deer, 1,089 roe deer, 611 white-tailed deer, 262 moose, 165 fallow deer, 30 reindeer, 2 wild forest reindeer (Rangifer tarandus fennicus), 1 sika deer and 560 deer of unknown species. No TSE-positive cases were detected1-20).
I t was concluded that the results indicated that there was not a TSE epidemic in cervids in the EU. However, the survey had several limitations. These included, among others, difficulties in the collection of samples (not all member states achieved the required sample size), the limited number of potentially susceptible cervids tested, the fact that the sampling could not be assumed to be random and that the testing had been done in brainstem only (not including lymph node), which may have affected the sensitivity of detection. Owing to the limitations explained above, and also taking into account that CWD spreads from and within clusters and that CWD susceptible cervids are present in Europe, it was concluded that occurrence of cases of TSE, particularly in remote areas and areas not sampled in the survey, may not be excluded (20).
Few other large-scale studies on the occurrence of CWD/ TSEs in cervids have been conducted in Europe. A large survey was done in Germany from 2002 to 2005. In total, 7,056 wild cervids (red deer, fallow deer and roe deer) and 248 farmed cervids were tested with a rapid test on samples of brainstem (obex) and medial retropharyngeal lymph nodes. Eighty-seven per cent of the administrative districts in Germany were sampled, including different hunting areas and including as many deer populations as possible, attempting to identify potential clusters of CWD. No positive results were found, and it was concluded that TSE was unlikely to occur in free-ranging cervids in Germany, and if it occurred it was not extensively distributed(23).
TRANSMISSION
BSE is transmitted by the oral route. Horizontal or vertical transmission has not been detected; however, the offspring of clinical cases of BSE have shown an increased risk for BSE.
Scrapie is maintained and disseminated by lateral transmission. PrPSc may be demonstrated in the placenta of infected ewes, which is a source of infection for the offspring and other sheep in the flock.
CWD is contagious and is transmitted horizontally and through the environment. Pastures and enclosures contaminated by saliva and excreta and by carcasses of animals that died from CWD are the primary source of infec- tion(24). PrPCWD binds with high affinity to clay constituents of soil and contamination persists for years(25). PrPCWD has also been demonstrated in water and can even be transmitted through bedding material and water from pens used by infected animals(26). CWD is also transmitted by direct contact and transmission can occur among deer of the same species and of different species. CWD is easily transmitted among captive deer and elk concentrated in enclosures, and it is believed that artificial feeding and baiting facilitates transmission by concentrating animals.
In atypical scrapie and atypical BSE there are no indications of transmission. Only single sporadic cases with a wide geographical distribution occurred.
Pathogenesis and pathology
TSE in animals are mainly acquired by oral infection. During the early phase, PrPSc crosses the mucous membranes and is deposited in the tonsils, Peyer’s patches and lymph nodes draining the gastrointestinal tract. Experimentally, the early involvement of lymphoid tissue in CWD has been observed as early as 6 weeks post- infection(27) and in natural scrapie infection in lambs from 3 months of age. PrPSc/PrPCWD replicates in the lymphore- ticular system, including the spleen and lymph nodes, during a prolonged phase of months to years. In TSE the kinetics of the infection and distribution of prions is affected by the host species and its Prnp genotype. In BSE and in some scrapie cases there is little involvement of the lymphoreticular system.
At later stages of TSE infection, PrPSc extends to the brain, where it progressively accumulates, causing fatal neurodegenerative changes. The mechanisms of transit of the PrPSc from the gastrointestinal tract or tonsils into the brain are not yet fully understood. Two routes of transit have been implicated: the haematogenous route and the retrograde axonal routes, involving fibres innervating lymphoid tissues or the autonomic nervous fibres of the digestive tract. The parasympathetic dorsal motor nucleus of the vagus nerve (DMNV) is the site of earliest involvement in the brain in scrapie and CWD and occurs before onset of clinical disease. In BSE, the first site of prion accumulation in the brain is the nucleus of the solitary tract. Both the DMNV and the solitary tract nuclei are located in the caudal brainstem, at the level of the obex. From these early sites the infection extends to other parts of the brain, targeting specific locations, which depend on the type of TSE, host species, genotype and prion strain.
During late phases of infection, PrPCWD is extensively distributed in the body and is present in tonsils, spleen, retina, skeletal muscle, fat, peripheral nervous system, adrenal glands, blood, saliva, lymph nodes, brain and spinal cord and antler velvet. PrPCWD is shed in faeces, urine and saliva, mainly during terminal disease1-28’29).
The TSE are characterized by two types of vacuolar lesions in the grey matter, affecting specific target nuclei/ areas. One is vacuolation of neuronal processes, called neuropil vacuolation or spongiosis, and the other is intraneuronal vacuolation. The PrPSc accumulation in the brain is characteristic of TSE and can be detected at earlier stages than vacuolar lesions. No inflammatory reaction and no immune response appear to be developed against the pathogenic PrP, presumably because it is not perceived as foreign by the immune system. Amyloid plaques may occur in some TSE. No histological lesions are observed in lymphoid tissues in any of the TSE, even with advanced PrPSc accumulation.
CLINICAL SIGNS AND TREATMENT
In general, TSE are clinically manifested by neurological signs and loss of body condition.
Clinical signs of CWD characteristically appear after 1.5 years of age and are most frequent in animals 3 to 5 years old. The signs include behavioural changes, such as depression, aggression, somnolence and separation from other animals; signs of neurological disturbance such as paralysis, dysphagia, hind limb ataxia, head tremors and recumbency. Other signs are increased salivation, progressive weight loss, polyuria and polydypsia, and signs related to aspiration pneumonia(15).
CWD has a long incubation time; following oral experimental infection, the average time from infection to death is 23 months (with a range of 20—25 months). Most animals that test positive to CWD do not exhibit clinical signs(15).
DIAGNOSIS
The selection of diagnostic techniques for TSE depends on the purpose of the testing: screening, confirming TSE or typing a strain. For CWD it was stated by the US Department of Agriculture that the available tests could only be used as a surveillance tool and not for food safety purposes. Similarly, the EU has also approved TSE tests only for surveillance. Current TSE tests are not considered to be suitable for food safety control, because there is not enough knowledge on the infectious dose for natural TSE in animals and humans, on the PrPSc tissue distribution and infectivity, and on the stage in the incubation period at which the tests are able to detect natural infection.
The post mortem diagnosis of well-established TSE poses no major difficulties. However, tests applied to cases with minimal accumulation of PrPSc or at just about the detection limits of the test, such as during early-stage infection, may give false negative or inconclusive results. Screening tests with high sensitivity may either detect such cases as positive or may classify them as ‘suspects’, and their confirmation with another test may be more difficult and may result in discrepancies in results. Different test results may also derive from differences in protocols, especially for inhouse developed IHC and WB techniques. Commercial tests for TSE have been validated and are officially approved. The EFSA evaluated data provided by companies producing tests for diagnosis of CWD and deduced that all the tests evaluated would be suitable for surveillance in Europe(30). Direct comparisons of sensitivity and specificity of the tests are often not correct because of the high variation in the design of the validation studies.
DIAGNOSTIC SAMPLES
PrPSc accumulates in lymphoid tissues in TSE in deer, North American elk, sheep and goats at earlier stages than in the brain; therefore, sampling lymphoid tissue allows detection of early infection. However, in some cases, mostly in elk and in sheep with genotypes with no peripheral PrPSc accumulation, PrPSc/PrPCWD is detected by IHC in the brainstem but not in single sections of either tonsils or retropharyngeal lymph nodes. This is probably due to the lower levels of accumulation of PrPCWD in lymphoid tissues in early phases of infection, often focal and restricted to a few follicles. Therefore, the highest sensitivity of detection of CWD is achieved by testing both lymphoid tissue and brainstem at the level of the obex.
Brain
The medulla oblongata at the level of the obex is an optimum sample for diagnosing BSE, CWD and typical
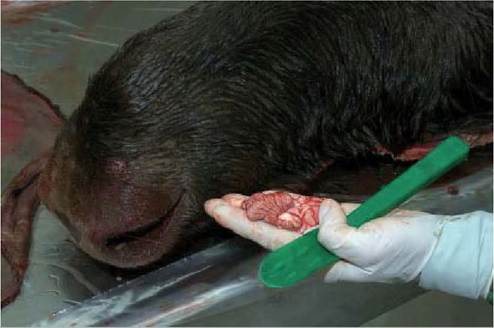
FIGURE 43.1 Sampling of a moose for testing for CWD. A sample of brainstem (a piece of cerebellum is left attached) was obtained by introducing a disposable instrument in the foramen magnum. Photo: Bengt Ekberg, SVA.
scrapie because of its consistent early accumulation of PrPSc and vacuolar lesions. For BSE an appropriate sample for diagnosis should include the solitary and the trigeminal tract nuclei. For CWD and scrapie it is critical that the DMNV is represented, particularly to detect early or sub- clinical infections. For atypical scrapie, the trigeminal tract nuclei and particularly the cerebellum are required for confirmation. Brainstem samples are conveniently obtained by introducing a commercially available long spoonshaped metal or disposable instrument with cutting edges, through the foramen magnum (Figure 43.1).
Lymphoid Tissues
Because of the convenience for sampling and the close association with the gastrointestinal tract, the medial retropharyngeal lymph nodes, the tonsils and rectal biopsies are the lymphoid tissues of choice for testing for CWD and scrapie. As PrPSc accumulates in lymphoid follicles, it is important that they are well represented in the sample; for example, in lymph nodes the cortex (which contains follicles) has to be included.
Testing tonsillar or rectal biopsies allows the ante mortem diagnosis of CWD and scrapie, even in pre-clinical stages of infection. The methods to obtain rectal biopsies are well developed, easily performed and allow sampling of several biopsies over different time intervals, without causing significant damage to the mucosa. Special disposable instruments are commercially available to obtain the rectal biopsies. As the number of lymphoid follicles in the recto- anal lymphoid tissues decreases with age, the diagnosis performs best in elk younger than 8.5 years(31).
Autolysis
The PrPSc is very resistant to degradation, and its detection is not significantly compromised by autolysis. However, the early accumulation of PrPSc in the brain is very restricted neuroanatomically, and the biggest threat to sensitivity in the autolysed tissues is the uncertainty as to whether the target areas were actually tested.
Diagnostic Methods
Most of the diagnostic methods for PrPSc detection in tissues rely on its protease resistance and identification by PrP antibodies.
To date, there are no reliable non-invasive tests to diagnose TSE in live animals. Progress has been made on the development of in vivo tests for urine or blood, but sufficient sensitivity and consistency have not yet been obtained. Even though some tests for live animals are already commercially available their performance is not yet well assessed. As immunological response to PrPSc is not developed, immunological tests cannot be used.
Rapid tests or screening tests are tests of high sensitivity, which can be completed usually in a few hours, and are suitable for high throughput testing, for example, for testing hunter killed populations. Most of them are based on enzyme-linked immunosorbent assay (ELISA) or WB methods, and many are commercially available, validated and approved for screening (see information about their application in the survey section above).
Confirmatory tests are tests of high specificity used to confirm clinical suspicion of disease, or positive or inconclusive rapid test results. In most countries, only the confirmed BSE and scrapie cases are considered to be positive. The methods that are recommended as confirmatory tests consist of histopathology, IHC, detection of scrapie- associated fibril (SAF) and WB.
Immunohistochemistry
IHC recognizes the presence of distinctive PrPSc deposition in the target areas or cells and is therefore the most specific test for TSE. IHC is usually done on formalin- fixed samples. It applies a pre- treatment of the sections with formic acid and hydrated autoclaving for epitope demasking and to remove the normal PrPC. Numerous anti- PrP monoclonal and polyclonal antibodies have been developed and many are commercially available. IHC has also been used as a screening test for CWD in automated high output systems. For brainstem samples, the target nuclei have to be well represented and recognized in the section. The early distribution of PrPSc in the lymphoid tissue is not homogenous; therefore, it is considered that when applying IHC a minimum number (usually 4 to 6) of follicles with germinal centres need to be examined to provide a reliable negative diagnosis.
I n the early days histopathology and the detection of SAF by negative contrast electron microscopy were the only available tests for diagnosis of TSE. Today SAF is rarely used because of its low sensitivity compared with the rapid tests, but it may be a method of choice when only very autolysed or only formalin-fixed sample is available.
Strain Discriminatory Methods
In vitro methods to typify prion strains have been developed to differentiate BSE from scrapie in sheep. A recent study, albeit on a small number of samples, showed that it was also possible to discriminate between CWD in North American elk and white-tailed deer, and BSE and scrapie cases(32). The methods are based on the different sensitivity of the prion strains to PK digestion. The precise basis for discrimination is the location of the N-terminal cleavage site for proteinase K digestion of PrPres between BSE and scrapie. Although in vitro methods give a preliminary molecular description of the isolate, they cannot actually identify a strain. The ultimate typing of the prion strain is still determined by bioassay, most commonly in mice, as molecular characterizations are not fully specific.
MANAGEMENT, CONTROL
AND REGULATIONS
The BSE concern prompted large-scale testing in Europe and elsewhere. For a prolonged period, European regulations required rapid screening for BSE on all cattle aged over 30 months slaughtered for human consumption. More than 87 million cattle have been tested since 2001. The TSE control measures implemented in the EU resulted in a marked decline in the number of cases and the regulations have been adapted accordingly for example by increasing the age limit of obligatory TSE testing of cattle to 72 months.
With the application of TSE screening rapid tests to large-scale testing of animals, and a battery of confirmatory and strain- typing methods, new atypical forms of TSE were identified in sheep and cattle. It is hypothesized that atypical TSE may represent spontaneous forms of disease related to old age, caused by mutations in the Prnp gene equivalent to sCJD in humans, rather than prion infections contracted horizontally or orally. It can be speculated that such spontaneous diseases could occur in other species, including cervids and other wild animals. If any type of prion disease is detected in a wild animal in Europe it will need to be confirmed and fully characterized. The confirmation is done by the EU reference laboratory for TSE(33).
In North America, surveillance of CWD includes targeted surveillance, testing of cervids with clinical suspicion, and hunter harvest- based surveillance — testing of healthy animals done on the heads submitted voluntarily by hunters. Additionally, road- killed cervids and cervids found dead are tested in parallel. CWD is contagious, is also transmitted through environmental contamination and migrates over large distances, so its eradication from free-living cervids is considered unlikely.
PUBLIC HEALTH CONCERN
The finding that vCJD, a new variant of CJD, was associated with PrPBSE was alarming. More than 160 cases of vCJD have been diagnosed in the UK and smaller numbers in other countries. The public fear increased when likely iatrogenic vCJD transmission via blood products was reported(34). However, as the EU-wide control strategies were successful and the number of BSE cases in Europe has decreased drastically, the risk for humans has also decreased considerably.
Fortunately, BSE in small ruminants was found to be only a rarity and therefore potential infectivity to humans of BSE from species other than cattle is no longer of major concern. The infectivity for humans and other animals of the atypical strains of TSEs in cattle and sheep is not fully established.
The BSE experience and the unknowns about TSE resulted in concerns about a potential food-borne zoonotic role for other prion diseases of animals, and CWD was an example. CWD as a possible cause of CJD has been questioned, but fortunately, a higher incidence of CJD in hunters, their relatives and venison consumers has not been found. It is reassuring that until today, no human TSE infection acquired through consumption of venison or sheep meat has been shown. Still, caution through human avoidance of consumption of TSE-infected products is recommended(35).
REFERENCES
1. Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136-44.
2. Advisory Committee on Dangerous Pathogens. Categorisation of Biological Agents According to Hazard and Categories of Containment, 4th edn. Sudbury, UK: HSE Books; 1995.
3. Benestad, S., Sarradin, P, Thu, B. et al. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Veterinary Record. 2003;153:202-8.
4. Benestad, S., Arsac, J.N., Goldmann, W. & Noremark, M. Atypical/ Nor98 scrapie: properties of the agent, genetics, and epidemiology. Veterinary Record. 2008;39:19.
5. Wells, G., Scott, A.C., Johnson, C.T. et al. A novel progressive spongiform encephalopathy in cattle. Veterinary Record. 1987;121:419-20.
6. Kirkwood, J., Cunningham, A.A. Epidemiological observations on spongiform encephalopathies in captive wild animals in the British Isles. Veterinary Record. 1994;24:296-303.
7. Sigurdson, C. & Miller, M.W Other animal prion diseases. British Medical Bulletin. 2003;66:199-212.
8. Willoughby, K., Kelly, D.F., Lyon, D.G. & Wells, G.A.H. Spongiform encephalopathy in a captive puma (Felis concolor). Veterinary Record. 1992;121:431-4.
9. Wyatt, J., Pearson, G.R., Smerdon, T etal. Naturally occurring scrapielike spongiform encephalopathy in five domestic cats. Veterinary Record. 1991;129:233-6.
10. Bellworthy, S., Hawkins, S.A., Green, R.B., et al. Tissue distribution of bovine spongiform encephalopathy infectivity in Romney sheep up to onset of clinical disease after oral challenge. Veterinary Record. 2005; 156:197-202.
11. Rules for the prevention, control and eradication of certain transmissible spongiform encephalopathies (and amendments). Regulation (EC) No 999/2001: 2001.
12. Eloit, M., Adjou, K., Coulpier, M. et al. BSE agent signatures in a goat. Veterinary Record. 2005;156:523^.
13. Williams, E. & Young, S. Chronic Wasting Disease of captive mule deer: a spongiform encephalopathy. Journal Wildlife Diseases. 1980;16:89-98.
14. Sigurdson, C. A prion disease of cervids: chronic wasting disease. Veterinary Research. 2008;39:39-41.
15. Williams, E. Chronic wasting disease. Veterinary Pathology. 2005;42: 530-49.
16. Balachandran, A., Harrington, N.P, Algire, J. et al. Experimental oral transmission of chronic wasting disease to red deer (Cervus elaphus elaphus}: early detection and late stage distribution of protease-resistant prion protein. Canadian Veterinary Journal. 2010;51:169-78.
17. Dagleish, M., Martin, S. & Steele, P. Experimental transmission of bovine spongiform encephalopathy to European red deer (Cervus elaphus elaphus}. BMC Veterinary Research. 2008;4:17.
18. Martin, S., Jeffrey, M., Gonzalez, L. et al. Immunohistochemical and biochemical characteristics of BSE and CWD in experimentally infected European red deer (Cervus elaphus elaphus). BMC Veterinary Research. 2009;5:26.
19. Peletto, S., Perucchini, M., Acin, C., et al. Genetic variability of the prion protein gene (PRNP) in wild ruminants from Italy and Scotland. Journal of Veterinary Science. 2009;10:115-20.
20. European Food Safety Authority. Scientific opinion on the results of the EU survey for Chronic Wasting Disease (CWD) in cervids. EFSA Journat. 2010;8:1861.
21. Opinion of the Scientific Panel on biological hazards (BIOHAZ) on a surveillance programme for Chronic Wasting Disease in the European Union. 2004, EFSA Panel on Biological Hazards. EFSA Journal doi:10.2903/j.efsa.2004.70.
22. Baeten, L.A., Powers, B.E. & Jewell, J.E. A natural case of chronic wasting disease in a free-ranging moose (Alces alces shirasi). Journal of Wildlife Diseases. 2007;43:309-14.
23. Schettler, E., Steinbach, F., Eschenbacher-Kaps, I. et al. Surveillance for prion disease in Germany. Emerging Infectious Diseases. 2006;12:2.
24. Miller, M.W., Williams, E.S., Hobbs, N.T. & Wolfe, L.L. Environmental sources of prion transmission in mule deer. Emerging Infectious Diseases. 2004;10:1003-6.
25. Johnson, C.J., Phillips, K.E., Schramm, PT., McKenzie, D., Aiken, J.M. & Pedersen, J.A. Prions adhere to soil minerals and remain infectious. Plos Pathogens. 2006;2:296-302.
26. Mathiason, C.K., Hays, S.A., Powers, J. et al. Infectious prions in pre- clinical deer and transmission of chronic wasting disease solely by environmental exposure. Plos One. 2009;4:e5916.
27. Sigurdson, C.J., Williams, E.S., Miller, M.W et al. Oral transmission and early tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). Journal of General Virology. 1999;80:2757-64.
28. Haley, N.J., Seelig, D.M., Zabel, M.D., Telling, G.C. & Hoover, E.A. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. Plos One. 2009;4:e4848.
29. Tamguney, G., Miller, M.W, Wolfe, L.L. et al. Asymptomatic deer excrete infectious prions in faeces. Nature. 2009;461:529-U90.
30. European Food Safety Authority. Available online at: http://www.efsa. europa.eu∕en∕efsajournal∕pub∕70.htm [accessed 26 March 2012].
31. Wolfe, L., Spraker, T.R., Gonzalez, L., et al. PrPCWD in rectal lymphoid tissue of deer (Odocoileus spp.). Journal of General Virology. 2007;88:2078-82.
32. Stack, M.J., Balachandran, A., Chaplin, M. & Davis, L. The first Canadian indigenous case of bovine spongiform encephalopathy (BSE) has molecular characteristics for prion protein that are similar to those of BSE in the United Kingdom but differ from those of chronic wasting disease in captive elk and deer. Canadian Veterinary Journat. 2004;45: 825-30.
33. EU reference laboratory for TSE. Available online via: http:// vla.defra.gov.uk/vla/vla_aboutvla.htm [accessed 26 March 2012].
34. Llewelyn, C.A., Hewitt, PE., Knight, R.S. et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417--21.
35. WorldHealthOrganization. WHO Guidelines on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies. Geneva: WHO; 2006.