Inferences on the Origin of M. bovis
This riddle cannot be solved without revisiting the turbulent history of humans on the African continent. The data used are not exhaustive (not all countries in Africa are represented), but it is the most comprehensive collated data set to date using three genotyping tools as discussed earlier (Sect.
8.4).In this section, we circumstantially link observed molecular marker patterns of M. bovis to events during three periods in the history of the African continent: the precolonial, colonial, and postcolonial eras.
8.6.1 IndigenousAfricanStrains
Archaeological and osteo-forensic studies show that the Shurmuk and Uyuk people who occupied the southern Siberian region during the second century BC (~4000 years ago) were plagued by tuberculosis caused by M. bovis (Taylor et al. 2007). Based on this report, if we assume that cattle at that time were infected with M. bovis, and that they were the most probable source of this infection in the Shurmuk and Uyuk communities, then it is logical to assume that the disease was not limited to that region but that it could have been present in any settled community that was rearing cattle at that time. Donoghue (2009), using ancient biomolecules to study the MTC, concluded that this human infection could have been the consequence of prolonged close interaction between these semi-nomadic pastoralists and their cattle. If this were indeed the case, then the transhumant communities of Northern Africa, the Bantu, Luo, Mande, and the Omotic people who lived at the same and more recent times (Clark and Brandt 1984) could have farmed with cattle that were infected with M. bovis.
There is a small, but unique, collection of spoligotypes that are only found in Africa (Muwonge et al. 2016). This suggests that they are most likely genotypes that were native to Africa and that they were present before European settlement and the importation of M.
bovis-infected cattle. These spoligotypes include SB1100 and SB1102 from Chad (Muller et al. 2008); SB1003 and SB1200 from Tunisia (Ben Kahla et al. 2011); SB1265, SB1476, SB1517, and 1520 from Ethiopia (Biffa et al.2010) ; SB1405 and SB1406 from Uganda (Oloya et al. 2007); and SB1536, SB1572, and SB1765 from Zambia (Munyeme et al. 2009b).
8.6.2 Introduced Strains
It has been assumed for some time that M. bovis was introduced onto the African continent during the European colonization (Muller et al. 2009; Berg et al. 2011; Smith et al. 2011). Given the detection of the unique African strains, it is likely that M. bovis infections predate the European presence on the Africa continent. It seems to be more likely that the colonial era saw an introduction of new (additional) strains that influenced the evolutionary course of M. bovis as reflected by the variety and distribution of the strains that are currently present. These introductions during the colonial times are likely to have occurred in four ways:
1. The introduction of large numbers of cattle by European settlers following the colonization of South Africa (and other African countries)
2. By the importation of bulls for breed improvement purposes during the colonial and postcolonial times (Armando 1995; Taneja 1999; Stackyard 2009)
3. The introduction of exotic female cattle for commercial dairy farming (colonial and postcolonial era) (Stackyard 2009)
4. For restocking the decimated African cattle populations following the rinderpest epidemic during the course of and until the end of the nineteenth century (Bienart 1989; Mariner et al. 2012)
The introduced genotypes (spoligotypes) are likely to have the following characteristics: They are present in both colonial and colonized territories, and they are the dominant spoligotypes given that they are from regions with intensive husbandry systems (maximum number of animals per unit area) and intensive disease control (test-and-slaughter) strategies.
These genotypes originated from a system in which there was increasing transmission pressure and a population bottleneck and had to be better adapted (Smith et al. 2006). For example, France and Britain started their attempts to eliminate BTB in the 1930s at the height of their colonial power (More and Good 2006; Smith et al. 2006; Berdah 2010). These interventions were increased during the 1950s and up to the 1980s. The time could not have been better for these new M. bovis introductions as the African cattle herd was in the process of recovering from the population bottleneck caused by the rinderpest epidemic (Tambi et al. 1999; African Union 2010). This aggregate of factors likely created a conducive environment for the introduced new M. bovis strains to stamp their dominance wherever they were introduced. The current distribution of spoligotypes seemingly reflects this situation in that SB0944 is localized in Western Africa; SB0133 is prevalent in Eastern Africa; and SB0120, SB0134, SB0140, SB0121, SB0130, and SB0961 are found in Southern and Northern Africa (Muwonge et al. 2016). The most recent common ancestor (MRCA) of the clonal complexes documented in Africa (Muller et al. 2009; Berg et al. 2011; Smith et al. 2011) is also in this category, which corroborates this generalization in terms of origin.Clonal evolution is discussed in the next section (Sect. 8.6.3), but it may be argued that the African clones may have been introduced into Europe as some of the “spoils of the colonial conquest,” as has been suggested for other diseases (Jackson
2003). This notion, however, is at variance with the history of animal and human movements in Africa. In addition, SB0944 has only been reported in French cattle (Haddad et al. 2001) and livestock and humans in Francophone West Africa. It was reported outside this range only once in a man in Great Britain, whose origin was traced back to an Anglophone Western African country (Muller et al. 2009).
8.6.3 Understanding the Evolution and Phylogeny
of M.
bovisIn the past few decades, several robust molecular biological tools have been developed and applied in combination with conventional epidemiologic approaches. These have enabled researchers to carry out extensive, in-depth investigations to generate valuable information that enhanced the understanding of the molecular genetic characteristics of M. bovis. Such approaches revealed previously unknown genetic and microbiological characteristics of M. bovis, the genetic relatedness of the different types, and how the various genotypes with different virulence, and with geographic clustering, host preference, and adaptability evolved.
Subtractive genomic hybridization (Mahairas et al. 1996) revealed the genetic relatedness of the BCG vaccine strain with known virulent strains of M. bovis. The BCG strain is avirulent due to deletion of three genomic regions of difference (RD1, RD2, and RD3), suggesting a possible association between the evolutionary genomic conservation of genes existing in those regions and the extent of strain virulence. The loss of virulence by the BCG strain may be linked to the evolutionary loss of a gene regulatory function as a result of deleted RDs, particularly RD1. This finding paves the way for the possibility of developing new diagnostic tools to differentiate the immune response to BCG vaccination from infection with virulent strains (Mostowy et al. 2002).
The complete genomic sequence of M. bovis provided information about the evolution, molecular genetics, and phenotypic characteristics of the pathogen. The previously held dogma that M. bovis was the progenitor of the human pathogen, M. tuberculosis, was challenged by these data (Garnier et al. 2003). While M. bovis was initially assumed to have crossed the species barrier to the human host after the domestication of cattle, the deletion of certain genes from the genomic structure of M. bovis, which are present in M. tuberculosis, led to the current status where M. tuberculosis is believed to be closer to the ancestral progenitor of MTC than M.
bovis is (Brosch et al. 2002; Garnier et al. 2003). Further clarification of the variation in genomic characteristics of various M. bovis strains should assist researchers to understand how these phenomena influence the variation in virulence, host and geographic selection, and the adaptation of mycobacterial strains to host immune responses, and environmental pressures.8.6.3.1 Clonal Complexes
A clonal complex is defined as a group of strains belonging to abacterial species deemed to have descended from a single bacterial species cell (the MRCA) because they all bear characteristics derived from this single cell (Berg et al. 2011; Muller et al. 2009; Smith et al. 2011). In this section, we adapted this grouping, based on the findings of Berg et al. (2011), Muller et al. (2009), and Smith et al. (2011) who characterized M. bovis isolates from the African and European continents using spoligotyping and deletion analysis. There are currently three confirmed and two putative clonal complexes:
1. African 1 (Af1) is characterized by the genomic deletion of RDAf1, which is the absence of spacer 30 in the spoligotype pattern; SB0944 is the MRCA, and it is predominantly present in Western Africa.
2. African 2 (Af2) is characterized by the genomic deletion of RDAf2, which is the absence of spacers 3-7 in the spoligotype pattern; SB0133 is the MRCA, and it is predominantly present in Eastern Africa.
3. European 1 (Eur1) is characterized by the genomic deletion of RDEur1, which is the absence of spacer 11 in the spoligotype pattern, and it is predominant on the British Isles, but it is present worldwide.
4. Putative Af3 and Af5 clonal complexes are characterized, respectively, by the loss of spacers 3-5 and 3-5 and 8-10 in their spoligotype patterns and have been, respectively, reported in Mali and Burkina Faso, and Madagascar. The status of these two clonal complexes has not been fully confirmed, pending deletion analysis.
Given the strong correlation between spoligotype pattern, the region-of-differ- ence-based molecular markers (RDAf1, RDAf2, and RDEur1) (Muller et al.
2009; Berg et al. 2011; Smith et al. 2011), and geographical homology, spoligotypes in this section are discussed as pseudomolecular markers of evolution. This is based on the assumption that homoplasy is rare within a confined geographical area.8.6.3.2 West African Clonal Complexes
There are two clonal complexes in Western Africa: Af1 and the putative Af5 clonal complexes (Muller et al. 2009; Sanou et al. 2014). The former is the most dominant in this region, and spoligotype SB0944 is prevalent in all the countries in Western Africa. This spoligotype is central in the minimum spanning tree to which all the other spoligotypes in the region are rooted, and there is consensus that this pattern is the same as Af1's MRCA (Figs. 8.9 and 8.10). Although many countries have very similar spoligotype profiles, there are subtle country-specific differences, most of which will occupy less central positions (Fig. 8.9). To put it in numbers, about 90%- 95% of M. bovis isolates from Chad, Nigeria, and Cameroon belong to the Af1 clonal complex. In these three countries, SB0944 is the predominant spoligotype. However, other spoligotypes including SB1027 and SB1025 are frequently also
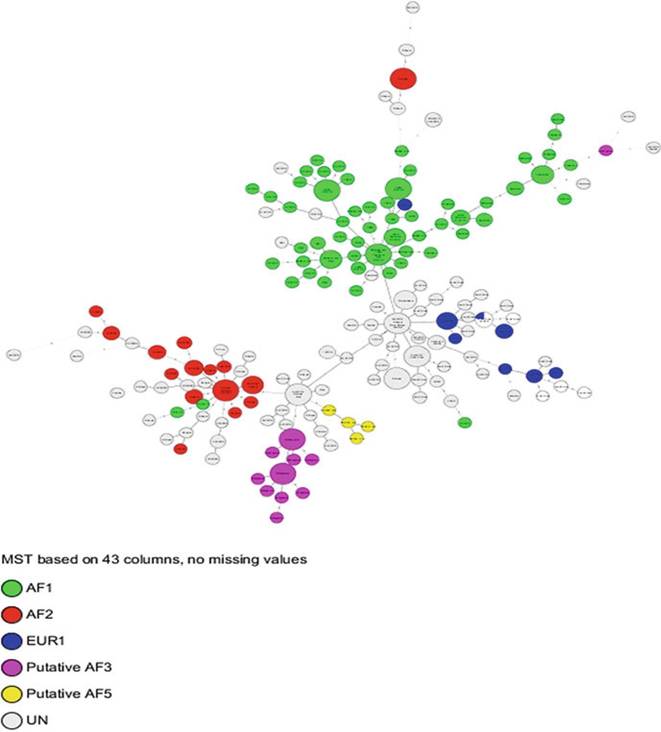
Fig. 8.9 African clonal complexes of M. bovis. (A) The minimum spanning tree (MST) was generated based on 43 spacer spoligotypes; each node of the MST represents a unique spoligotype, and the edge length and number represent the number of spacers deleted between any two connected nodes. UN denotes isolates that have not yet been assigned to a clonal complex
observed in Nigeria, but they are less common in Chad (Muller et al. 2009). Spoligotype SB0953, however, has been isolated exclusively in the Northwestern Region of Cameroon (Awah-Ndukum et al. 2013). Most of the spoligotypes that occur at low frequencies also tend to be country-specific (Figs. 8.11 and 8.12).
Another interesting feature of M. bovis isolates from West-Central Africa is the observed westwardly decreasing dominance of the Af1 clonal complex. Thus, only 76% and 60% of M. bovis isolates, respectively, from Burkina Faso and Mali belong to the Af1 clonal complex (Muller et al. 2008; Sanou et al. 2014). Unlike in the rest of the Western African countries, SB0300 that has only been reported in Burkina
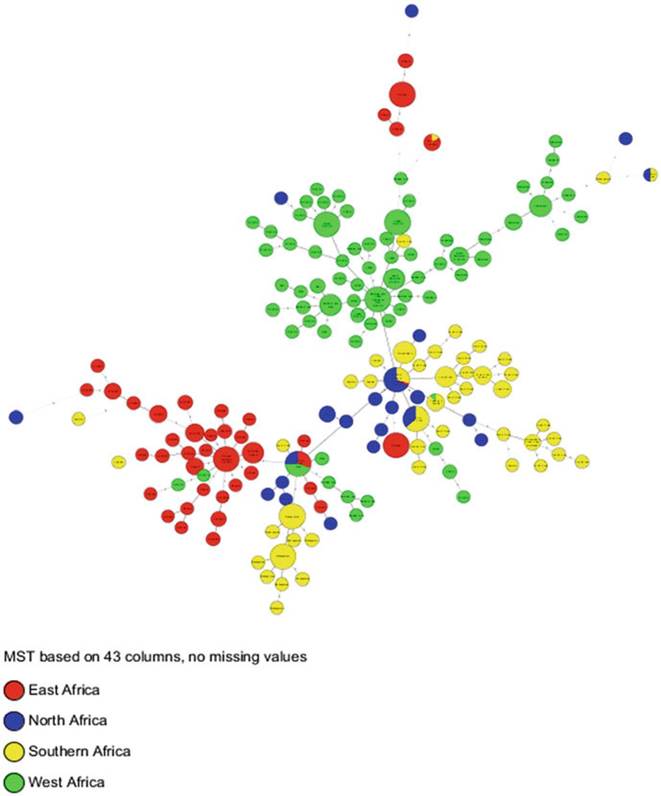
Fig. 8.10 Geo-distnbution of clonal complexes in Africa. The same parameters and calibration as in Fig. 8.9 were used, but they were color-coded by region
Faso and France (Haddad et al. 2001) is the most prevalent spoligotype in Mali. It is interesting to note that all spoligotypes, except for SB0944 from Mali, lack spacer 6 in their spoligotype pattern (Muller et al. 2008). The non-Af1 spoligotypes in these two countries belong to the putative Af5 clonal complex of which SB0134 is the most ubiquitous in Mali. The latter spoligotype has also been reported in Algeria (Sahraoui et al. 2009), Ethiopia (Biffa et al. 2010), France (Haddad et al. 2001), and Spain (Mateos et al. 1996).
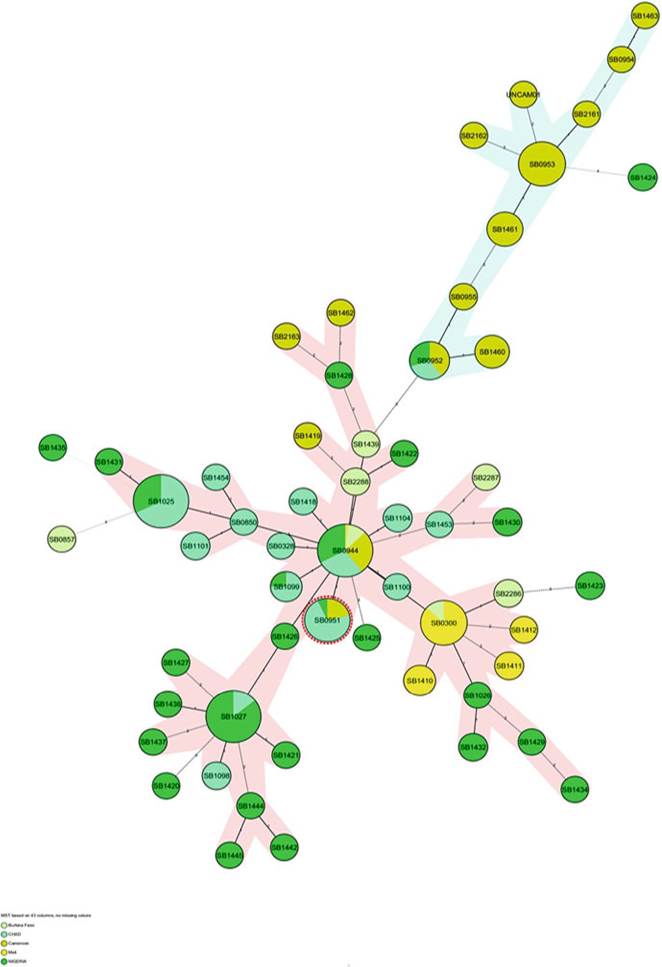
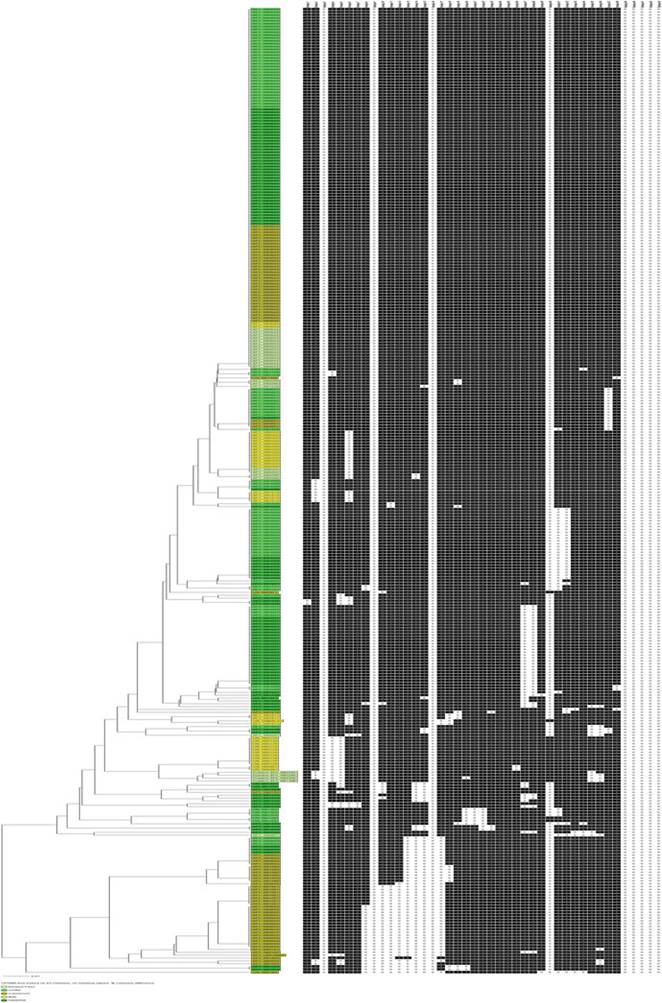
Fig. 8.12 Dendrogram showing the putative Af5 and unknown clonal complexes in five West African countries
8.6.3.3 East African Clonal Complexes
Af2 is the dominant clonal complex in East Africa. Unlike its Western African counterpart (Af1), where the MRCA is also the dominant spoligotype (SB0944), SB0133, Eastern Africa’s MRCA, is not the dominant spoligotype (Fig. 8.13) (Berg et al. 2011). This spoligotype has been recovered in samples collected from all Eastern African countries except from Burundi, although the results in Burundi are limited and are based on a single study that is not representative of the entire country (Rigouts et al. 1996; Berg et al. 2011). There is a score of non-Af2 isolates unique to Eastern African countries, such as SB1405 and SB1406 in Uganda, SB0425 in Tanzania, and SB1476 in Ethiopia. These have putatively been named the “indigenous east African spoligotypes.” Unlike in Western Africa, a BCG-like spoligotype has also been reported from Ethiopia (Biffa et al. 2010) and Zambia (Munyeme et al. 2009b).
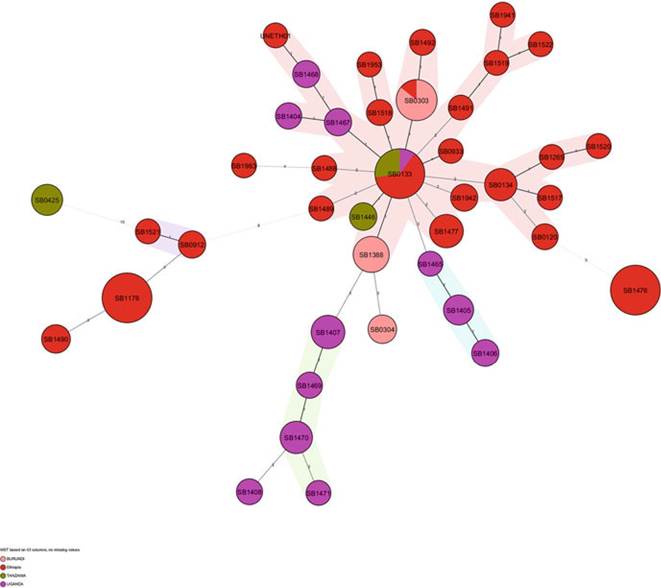
Fig. 8.13 The minimum spanning tree showing the distribution of clones (spoligotypes) belonging to the Af2 clonal complex in four Eastern African countries
8.6.4 Southern and Northern African Clonal Complexes
The spoligotype profiles of Southern and Northern Africa are very similar, although the regions are thousands of miles apart (refer to Sect. 8.6.2) (Figs. 8.14, 8.15, and 8.16). A significant proportion of the spoligotypes observed in these two regions belong to the Eur1 clonal complex, which is globally ubiquitous (Hlokwe et al. 2011; Sahraoui et al. 2009). This is specifically true for Algeria and South Africa, while a BCG-like spoligotype occurs in Zambia, and all the isolates from
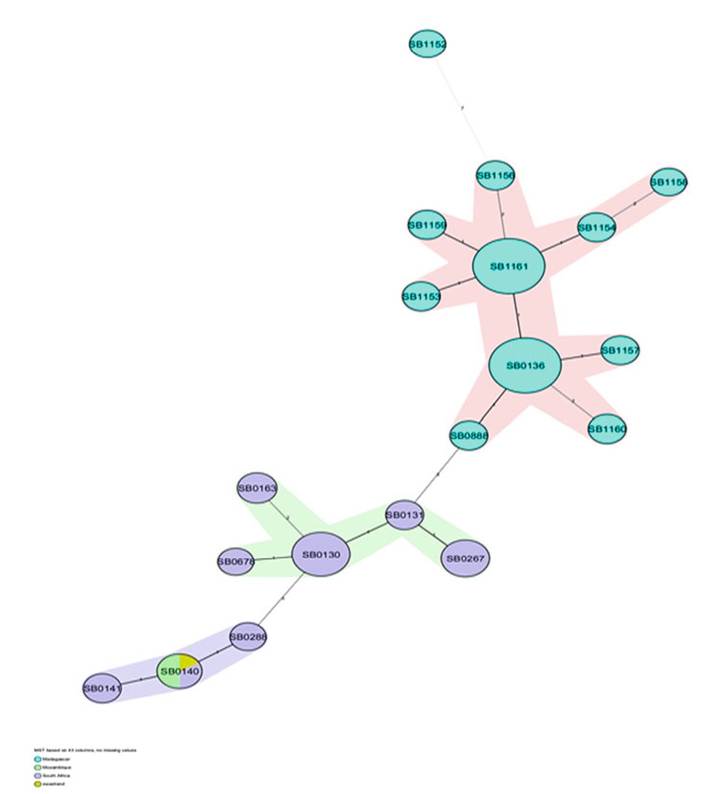
Fig. 8.14 The minimum spanning tree showing the distribution of clones (spoligotypes) belonging to the Eur1 and putative Af3 clonal complexes in four countries in Southern Africa
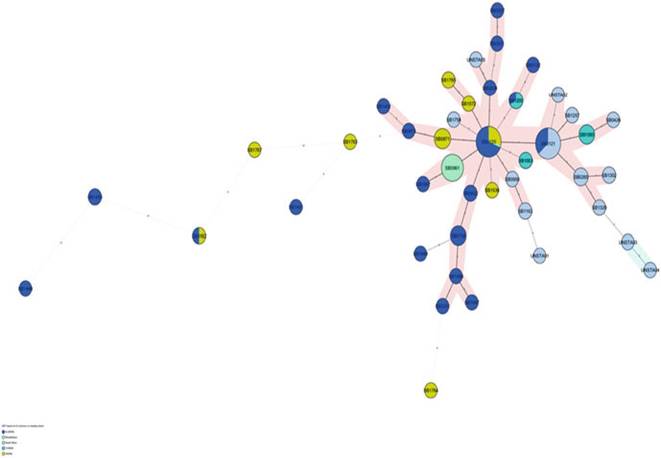
Fig. 8.15 The minimum spanning tree showing the distribution of clones (spoligotypes) that have not yet been assigned to a clonal complex in two Northern and three Southern African countries
Madagascar belong to the unique, putative Af3 clonal complex (Razanamparany et al. 2006; Muller et al. 2009; Munyeme et al. 2009b).
8.6.4 The Clonal Expansion and Spread of M. bovis
The various clonal complexes in Africa appear to be limited to specific geographical regions. The within-region distribution of the strains of specific clonal complexes, however, seems to be more complex. Although indigenous and introduced (exotic) genotypes determined the molecular landscape of M. bovis in Africa, it is more likely that the dynamics and practices of livestock movements, management, and trade had the biggest impact on the distribution of the various strains.
BTB was first documented in Cameroon in West Africa in 1913 when it was still a German-occupied territory. However, it was not uncommon to encounter BTB-infected cattle from Cameroon in Nigerian abattoirs in the 1940s, well into the time of French and British colonial rule (Alhaji 1976). These cross-boundary animal movements, driven by Sahel-West African transhumance, extended well beyond these two countries. The Sahel-West African transhumant movement is a giant carousel, involving the movement of between 70% and 90% of the regional cattle population, which provides 65% and 70%, respectively, of the meat and milk in the region (SWAC 2007). This cyclical animal movement occurs in the arid belt of
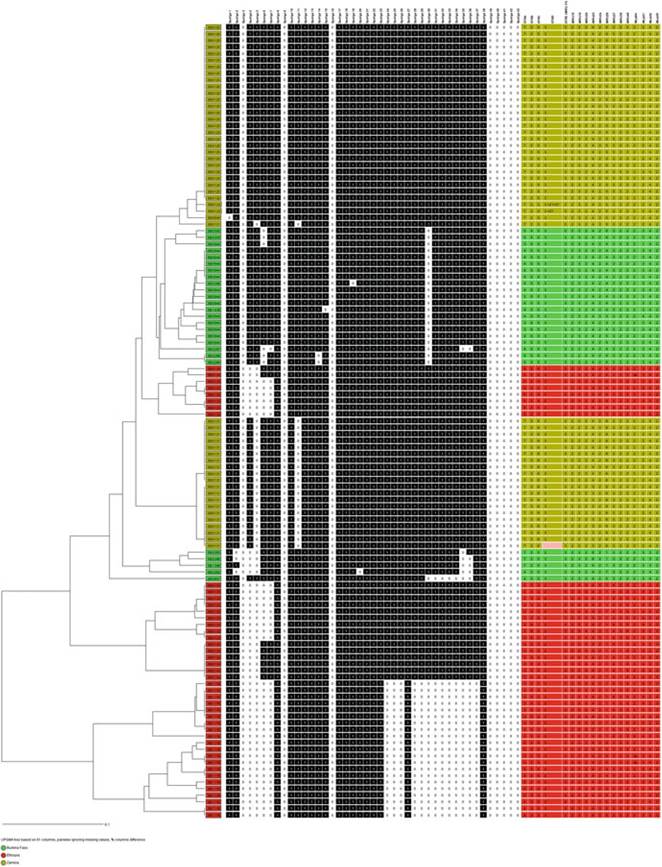
Fig. 8.16 A dendrogram showing diversity within selected spoligotypes in the east (Ethiopia in red), south (Zambia in yellow), and west (Burkina Faso in green) based on a MIRU-VNTR 18 loci panel using the Ridon version (Munyeme et al. 2009b; Biffa et al. 2014; Sanou et al. 2014)
Western Africa, involving half of Chad, Northern Cameroon, Nigeria, Burkina Faso, and most of Mali. Livestock system experts only recently acknowledged that this way of life was indeed profitable and competitive and that it played a pivotal role in maintaining the culture of the Peulh communities, who have existed in this way for thousands of years (Cour 2001; SWAC 2007). It is plausible that transhumance, the size of the cattle population, and marketing channels of beef and dairy cattle and their products were the likely mechanisms by which the Af1 clonal complex established itself in Western Africa. The granular movement of cattle in this giant carousel is, however, likely to be diffuse, and it is unlikely that cattle are herded over a distance of about 2800 km, from Kidal in Mali to Ngaoundere in Northern Cameroon, without encountering common sources of water and pastures and markets. These encounters from in-contact networks and herds from Ngaoundere are thus indirectly connected to Malian herds through degrees of separation while the cattle are herded through this great, arid expanse. The opposite is also true, and it is the likely explanation for the gradual decrease in the number of clones belonging to the Afl clonal complex, when moving west from Chad to Mali (Muller et al. 2008, 2009; Sanou et al. 2014). At a more granular level, there are no identical genotypes (combined spoligotypes and MIRU-VNTR) in Chad and Mali as opposed to the numerous shared clones in Mali and Burkina Faso and in Nigeria and Chad (Muller et al. 2008, 2009; Jenkins et al. 2011; Sanou et al. 2014).
In Eastern Africa, although the countries share a common MRCA (Berg et al.
2011), there is a preponderance of country-specific spoligotype profiles, suggesting that the Eastern African countries are not as connected as those in Western Africa. Ethiopia has the most elaborate animal movement network in Eastern Africa, and the country’s cattle movement network is connected to Eritrea, Kenya, and Sudan (see Chap. 14). No data exist for the M. bovis genotypes in Eritrea and Sudan, but it appears that Sudan is also connected to the Sahel-West Africa transhumance network (Fig. 8.1c), and although the Western and Eastern African clonal complexes appear to be limited to their respective geographical regions, Sudan may be the point of convergence of their distribution (Muller et al. 2009; Berg et al. 2011).
At a local level, Ethiopia has a centripetal animal movement network in which cattle filter through rural markets into the urban centers, especially Addis Ababa (Firdessa et al. 2012). Since most studies were conducted at abattoirs, the data reflect cross-sectional observations of the entire region rather than the molecular composition in the urban areas. The distribution of molecular markers in Ethiopia (Biffa et al. 2010, 2014; Gumi et al. 2012) and in Uganda (see Chap. 22) supports the existence of this centripetal movement. Recently, a centrifugal movement of cattle developed in Ethiopia where improved cattle breeds are moved from the urban dairies into rural areas as part of the dairy development program (Staal et al. 2008). This is likely to have a significant impact on the profile and distribution of genotypes of M. bovis and other disease agents in Ethiopia and other countries linked to its livestock movement network.
In Southern Africa, there is not an elaborate transhumance movement network as exists in Western Africa, but in recent years wildlife appear to be playing an increasing role in the epidemiology of M. bovis in the region (Michel et al. 2008; Munyeme et al. 2008; Hlokwe et al. 2011). Historically in Southern Africa, wildlife migration, prompted by seasonal changes that influence the abundance of water and pasture, occurred uninterrupted (de Garine-Wichatitsky et al. 2013). The pattern and
extent of this migration, however, changed and decreased over the past century mostly due to increasing livestock farming activities. Veterinary cordon fences erected to contain wildlife and the spread of transboundary animal diseases blocked these ancient migratory routes in countries such as Botswana, South Africa, and Mozambique (Gadd 2012). Contact between M. bovis-infected cattle and wildlife, especially lechwe (Kobus leche kafUensis) in Zambia and African buffaloes (Syncerus caffer) in South Africa led to spillover of BTB to wildlife in the absence, or well before the erection, of fences (Michel et al. 2008; de Garine-Wichatitsky et al. 2013).
During the last two decades, about 20 transfrontier conservation areas were established in sub-Saharan Africa. In addition, in South Africa the wildlife industry developed into the fastest growing agricultural sector, resulting in a rapidly expanding human-livestock-wildlife interface, and the associated risk of multidirectional transmission of diseases (Bengis et al. 2002; Cloete et al. 2007; De Garine-Wichatitsky et al. 2013). In South Africa, M. bovis has a broad host spectrum, and the disease is endemic in several of its ecosystems. Currently 21 different wildlife species are known to be infected. Although the majority of these are spillover hosts, which do not play a substantial role in the epidemiology of BTB, there is a risk of spillback to cattle, as was recently demonstrated (Musoke et al.
2015). There is an increasing risk to uninfected wildlife populations because of the translocation of M. bovis-infected, but undiagnosed wildlife at this expanding wildlife-wildlife interface in South Africa (Hlokwe et al. 2014).
These changes are likely to have an influence on the composition of molecular strains of M. bovis isolates in the following ways:
1. Cattle-specific genotypes of M. bovis transmitted to wildlife undergo clonal expansion in the wildlife populations in geographically restricted areas, with periodic spillover to other wildlife species. These genotypes characteristically have minimal diversity because of their restriction to these areas (Michel et al. 2008; Hlokweetal. 2011).
2. Genotypes established in wildlife following spillover from cattle increasingly spread between geographically linked wildlife populations, the rate of spread being a function of the extent of contact between these wildlife populations (De Garine-Wichatitsky et al. 2013; Hlokwe et al. 2014).
In Zambia’s Kafue flood plains, cattle herders still practice transhumance, and animals are moved to the lower plains at the commencement of the dry season in search of adequate pastures. These animals, tended by herdsmen, can remain in these areas for months-on-end depending on the length of the dry season. The majority of the people practicing this type of cattle grazing system are traditional pastoralists who lived in these areas for decades. Particularly in the Liuwa plains, a huge migration of wildebeest, the second largest migration of land mammals (the largest being in the Serengeti), takes place between Zambia and Angola (De Garine- Wichatitsky et al. 2013; Muma et al. 2013). In these events, the risk of spillback from infected wildlife to cattle and to the human populations enhances the risk of contracting the disease both for humans and animals and emphasizes the need to embrace the “One Health” approach in the control and management of BTB.
As evidence of the historical connectedness in Southern Africa, shared spoligotypes (SB0140 and or SB0131) occur in South Africa, Zambia, and Mozambique (Fig. 8.17). This may infer that they shared a common source of infection (Muller et al. 2009; Smith et al. 2011), that independent introductions into each of the countries occurred, or that a single introduction into one country then spread to the others due to the migration of M. bovis-infected wildlife. The role of wildlife in
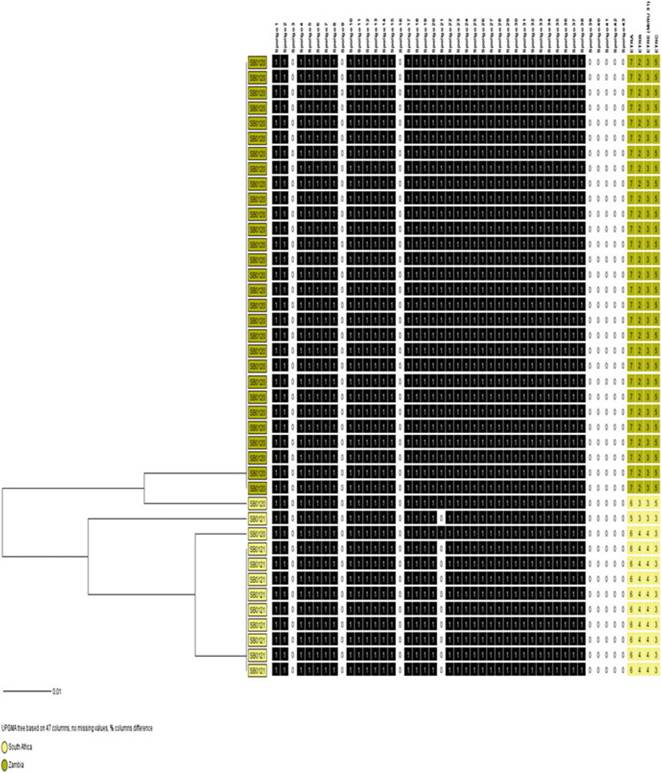
Fig. 8.17 Divergent evolution of SB0120 and SB0121 in South Africa (yellow) and Zambia (brown). The dendrogram was generated using spoligotypes and four exact tandem repeat loci (A-C and E)
this dissemination is currently speculative as it is not known whether they were infected with M. bovis at that time. However, when spoligotyping is combined with four loci of MIRU-VNTR for South Africa and Zambia (Figs. 8.18 and 8.19), it is evident that these apparently shared spoligotypes evolved divergently. If this situation also applies to countries such as Mozambique, Swaziland, and Botswana, it is possible that the restricted contact because of fencing, and international import control requirements that were more strictly applied on those countries, contributed to the divergent evolution of the strains in the respective countries. There are approximately 45, 42, and 73 unique M. bovis spoligotypes, respectively, in the Southern, Eastern, and Western regions of Africa, and the significant differences in
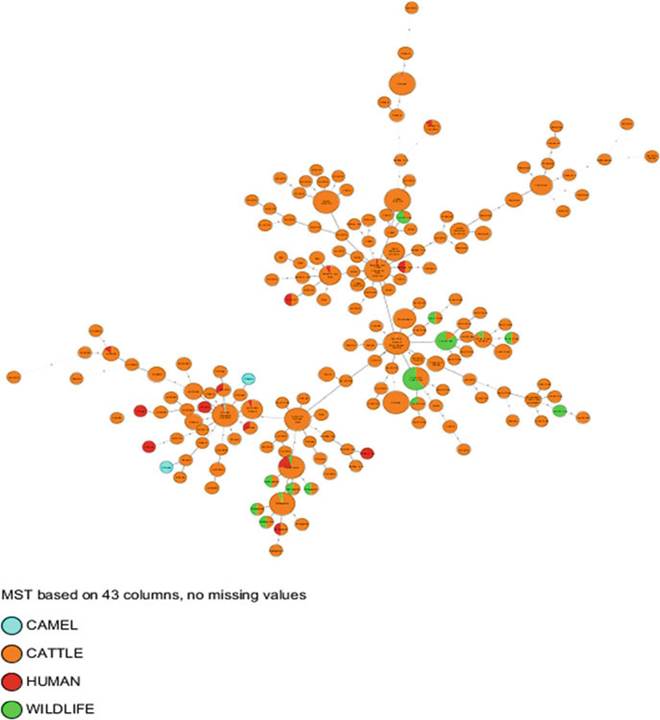
Fig. 8.18 The epidemiology of bovine and zoonotic TB in Africa: distribution according to host.
The MST was generated using the same parameters and calibration as in Figs. 8.9 and 8.10 but color-coded by host
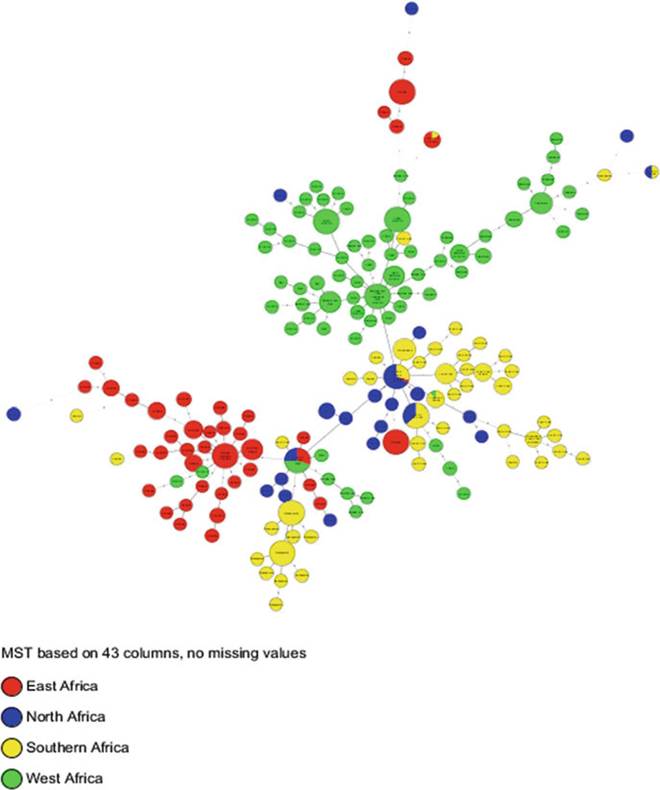
Fig. 8.19 The epidemiology of bovine and zoonotic TB in Africa: distribution according to country. The MST was generated using the same parameters and calibration as in Figs. 8.9 and 8.10 but color-coded by country
the spoligotype diversity between Western and Southern Africa support the bottleneck theory (Sect. 8.2.3). It is apparent from the molecular markers (Fig. 8.17) that countries such as Zambia and South Africa have had independent epidemiological dynamics for some time.
8.7