The Use of Molecular Epidemiology in Understanding the Dynamics of M. bovis
Molecular epidemiology is a field that emerged from the integration of molecular biology, clinical veterinary medicine, statistics, and epidemiology. It focuses on the role of genetic changes in organisms by linking their distribution within a population to events and niche-specific dynamics (Mathema et al.
2006). The data synthesis that underpins this multidisciplinary field is pivotal to the development and implementation of effective risk management and control strategies. This is especially so for a disease like BTB where a multidisciplinary approach to identifying disease determinants in human and animal populations is critical for resource-limited human and animal health systems.8.5.1 Understanding Genetic Diversity
Epidemiological studies in most countries are conducted to assess the genetic diversity and population structure of M. bovis. This information can then be used to explain the spread of a disease in different animal species. In Africa, these studies show varying levels of M. bovis diversity with the same typing tools (Oloya et al. 2007; Muller et al. 2008; Biffa et al. 2010; Hlokwe et al. 2011; Sanou et al. 2014). This reflects not only niche-specific genetic changes in pathogens but also hostpopulation dynamics given that pathogens are “hitchhikers” on the back of these events (Muwonge et al. 2016). This population-based diversity is a functional aggregate of molecular marker site diversity for the typing tool used and is assessed using the allelic diversity index. For example, the allelic diversity of MIRU-VNTR provides a measure of the discriminatory power of each locus and how it ultimately contributes to the total discriminatory power of MIRU-VNTR as a typing tool. The
Table 8.1 The discriminatory power of five exact tandem repeat (ETR) loci measured as the allelic diversity using 246 samples (Muwonge et al.
2016)| VNTR loci | Number of samples | Allelic diversity |
| ETRA | 246 | 0.780 |
| ETRB | 246 | 0.729 |
| ETRC | 225 | 0.543 |
| ETRD | 240 | 0.595 |
| ETRE | 246 | 0.254 |
Table 8.2 The
discriminatory power of
18 VNTR loci measured as the allelic diversity for
121 Mycobacterium bovis isolates (Muwonge et al. 2016)
| VNTR loci | Number of samples | Allelic diversity |
| ETRA | 121 | 0.710 |
| ETRB | 121 | 0.686 |
| ETRC | 121 | 0.581 |
| ETRD | 121 | 0.410 |
| ETRE | 121 | 0.041 |
| MIRU10 | 121 | 0.102 |
| MIRU16 | 121 | 0.554 |
| MIRU20 | 121 | 0.350 |
| MIRU23 | 121 | 0.204 |
| MIRU24 | 121 | 0.072 |
| MIRU26 | 121 | 0.729 |
| MIRU27 | 121 | 0.341 |
| MIRU39 | 121 | 0.257 |
| MIRU40 | 121 | 0.326 |
| Mtub04 | 121 | 0.104 |
| Mtub21 | 121 | 0.643 |
| Mtub30 | 121 | 0.391 |
| Mtub39 | 121 | 0.322 |
allelic diversity (h) for each locus is calculated using the formula (Selander et al. 1986):

where x is the frequency of the ith allele at the locus, n is the number of isolates in the analysis and (4ż) being the correction factor for bias in small samples.
Based on two databases (Muwonge et al.
2016), ETRA, ETRB, MIRU26, and Mtub21 are the most diverse allelic loci in Africa, which, by this measure, are the most desirable loci to include in a panel when MIRU-VNTR is used as a typing tool to investigate the diversity of M. bovis in Africa. ETRE, MIRU 24, Mtub04, and MIRU10 are the least allelic diverse loci, and the value of their inclusion in a panel to determine the diversity in M. bovis in Africa would be minimal (Tables 8.1 and 8.2).There are a number of methods available to determine spoligotype diversity (Biffa et al. 2010), but we evaluated it by assessing the number of unique spoligotypes detected in a specific country. Based on this approach, Nigeria, Ethiopia, South Africa, and Algeria have the highest spoligotype diversity (Fig. 8.8). At a
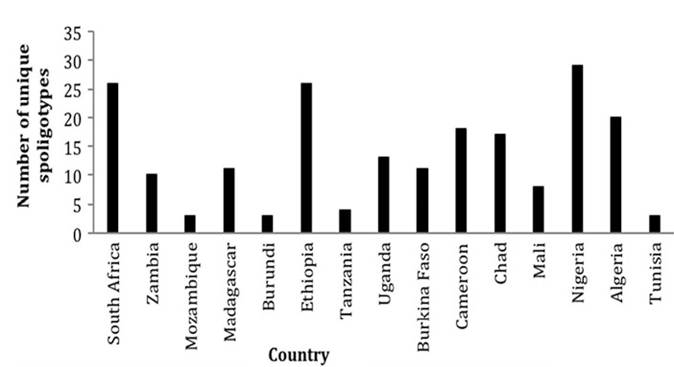
Fig. 8.8 Spoligotype diversity based on the number of unique spoligotypes recovered from each country in the last 20 years
regional level though, Western African isolates are almost twice as diverse as those present in Eastern and Southern Africa. The significance of this diversity will be discussed in Sect. 8.6.3.
8.5.2 Tracking the Sources and Routes of Transmission
In molecular epidemiology, transmission is inferred when homologous genotypes are recovered from two or more individuals linked spatially and temporally. These reflect genotypic clustering on phylogenetic visualization of isolates, and numerous molecular epidemiological studies in Africa reflect this attribute. For example, using clustering, in South Africa the increasing inter- and intraspecies transmission of BTB have been deduced based on this homology (Hlokwe et al. 2014). In Zambia, where transmission of M. bovis between cattle and lechwe (Kobus leche kafuensis) occurs, currently, lechwe are believed to be the largest BTB wildlife reservoir (Hang’Ombe et al. 2012). Similarly cross-species transmission has been inferred in Burkina Faso (Sanou et al.
2014), Ethiopia (Biffa et al. 2010), Madagascar (Razanamparany et al.2006), and Uganda (Muwonge et al. 2012; Oloya et al. 2008). Additionally, in Ibadan, Nigeria, there was homology between M. bovis genotypes isolated from slaughter cattle and from abattoir workers (Jenkins et al. 2011; Lawson et al. 2012).
In a wider geographical context, identical genotypes have been reported in Europe and Africa, especially in Northern and Southern Africa (Smith et al. 2011). The presence of these strains in these two African regions is not linked as their occurrence violates the space requirement for transmission. This matter will be dealt with in Sect. 8.6.3.
Source tracing is one of the aspects of disease investigation that benefits from the use of molecular epidemiological tools. This information can be beneficial in detecting on-farm sources of infection or contamination and tracing multiple-strain outbreaks (Biffa et al. 2010). The use of molecular markers to trace the sources of infections has been used in a number of other African countries including Madagascar (Razanamparany et al. 2006), Ethiopia (Biffa et al. 2010), Burkina Faso (Sanou et al. 2014), and Tanzania (Kazwala et al. 2001). In South Africa, for example, recent studies identified on-farm infections caused by two strains that were traced back to animals purchased and introduced from another farm (Michel et al. 2008; Hlokwe et al. 2014). Similarly, it has recently been shown that free- ranging African buffaloes play a vital role as a source of infection for livestock of neighboring communities in South Africa (Michel et al. 2009) and that wildlife to livestock spillover of BTB does occur (Musoke et al. 2015). In Zambia, infections in indigenous Zambian cattle in five of the six studied districts were traced to a common source (Munyeme et al. 2009a), while the zoonotic transmission of M. bovis in Namwala District has been traced back to cattle. Additionally, reverse zoonotic transmission of M. tuberculosis from humans to cattle has been detected in the same area (Malama et al.
2014). In Uganda, most zoonotic tuberculosis has been traced back to the local cattle populations (Oloya et al. 2007). Recently a potential alternative source of infection for humans has been identified in the form of spillover of the infection from cattle to pigs and then to humans (Muwonge et al. 2012).8.5.2 Elucidation of the Effect of Infection Pressure on the Severity of the BTB
Numerous studies have been carried out to determine the association between the virulence of genotypes of M. tuberculosis and the severity of the infection in humans. To date, there have been only a few studies conducted to characterize the severity of M. bovis infections in animals. The increasing availability of molecular tools in recent years allowed researchers not only to record the prevalence but also to describe the severity and extent of the lesions in infected herds. In this respect, molecular epidemiologic tools are used to demonstrate superseding genotype(s) in the area, the presence of polyclonality (simultaneous infection with multiple genotypes), and how these phenomena affect the severity of infection. Linking these data to spatial and temporal epidemiologic data allows researchers to understand the propagation of the infection and the persistence and evolution of the causal agent. For example, the combined use of MLVA and spoligotyping techniques provided an explanation of the increased severity of BTB lesions in Ethiopian cattle linked to a multiple-genotype infection and the spatial clustering of those genotypes. Among factors contributing to these phenomena were the rapid growth in livestock numbers, the expansion of intensive dairy farming, and the stock co-mingling, favoring the persistence and spread of infection in the area (Biffa et al. 2014). Similar studies in human populations in Uganda revealed a high prevalence of multiple-genotype/ strain/clonal infections in human TB patients (Muwonge et al. 2013), reaffirming the need for viewing tuberculosis lesions and the severity of clinical manifestations as highly dynamic processes that are more likely caused by coinfection with a number of strains in a single host.
8.6