Molecular Typing for Epidemiologic Studies of Mycobacterium bovis
Molecular epidemiology is distinct from the other subspecialties as it uses molecular biological tools to characterize nucleic acid- or amino acid-based content of microbial pathogens to study their distribution and the determinants of disease occurrence in host populations (Foxman 2001).
Typing is a process by which different organisms within a species are identified (Sabat et al. 2013). Originally this process was based on different phenotypes such as serotypes, biotypes, or antibiograms, but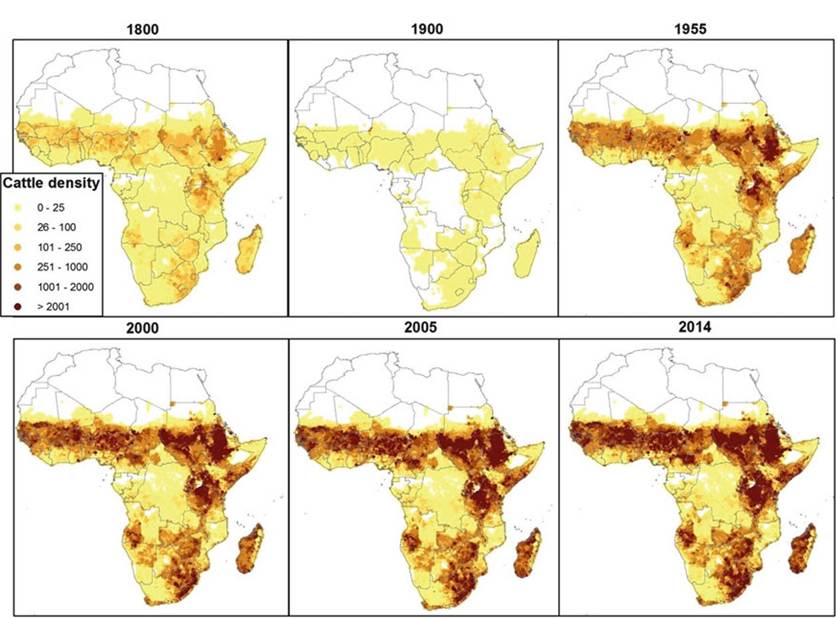
Fig. 8.3 Cattle population density in sub-Saharan Africa. The map was generated, using R and ArcGIS, based on population hind- and forecasting (FAO 2005), with 2005 as the point of reference
Molecular Epidemiology of Mycobacterium bovis in Africa
recent technological advances allow the use of molecular markers to determine the relatedness of isolates by using differences in conserved polymorphic genomic segments (Allix et al. 2004; Razanamparany et al. 2006; Sabat et al. 2013). Generally, the observed rate of the development of polymorphism (stability of the marker) and the genetic diversity of strains in the population are the key aspects when choosing an adequate molecular tool for studying the epidemiology of TB (Sabat et al. 2013). This implies that the rate of change of a marker must be sufficiently rapid to be able to distinguish between epidemiologically unrelated strains but slow enough to reliably link the related strains. These factors, together with the general background information of the prevalence of bovine tuberculosis, are normally taken into consideration when choosing molecular typing tools (Mathema et al. 2006).
The information in this chapter is based on the results of spoligotyping, MIRU- VNTR, and deletion analysis because they are the most commonly used techniques in Africa (Allix et al.
2004; Razanamparany et al. 2006; Sabat et al. 2013). In addition, spoligotypes are reported to mutate relatively slowly compared to the attributes detected by MIRU-VNTR that mutate relatively more rapidly (Reyes and Tanaka 2010). When used in combination with deletion analysis, the two techniques are suitable for examining the molecular epidemiology of BTB within historical and contemporary perspectives.8.4.1 Deletion Analysis
Screening for the deletion or presence of defined segments of DNA by Polymerase chain reaction (PCR) methods is widely used as a typing tool to identify different strains of the Mycobacterium tuberculosis complex (MTC). The presence or absence of region of difference (RD) has been used to infer the evolutionary process of members of the MTC of which M. bovis is a member. These host-specific strains arose from a common ancestor by the successive loss of segments (RD) of DNA (Fig. 8.4) (Brosch et al. 2002).
Mycobacterium bovis has further been classified into different clonal complexes based on deletion of specific, defined segments of its genome. Deletion of RDAf1, RDAf2, and RDEu1, respectively, characterizes the African 1 (Af1), African 2 (Af2), and European 1 (Eur1) clonal complexes, respectively, found in Western Africa, Eastern Africa, and the British Isles and their former colonies (Muller et al. 2009; Berg et al. 2011; Smith et al. 2011). These deletions are also highly correlated with the absence of spacers 30, 3-7, and 11 in Af1, Af2, and Eur1 spoligotypes, respectively.
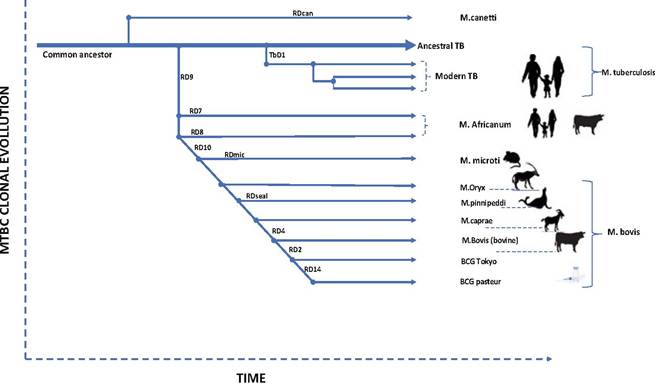
Fig. 8.4 Successive loss of DNA material. The scheme is based on the presence or absence of conserved deleted regions and on sequence polymorphism in five selected genes. The blue arrows indicate that strains are characterized by katG463 CTG (Leu) and gyrA95 ACC (Thr), typical for Group 1 organisms. The green arrows indicate that strains belong to Group 2 and are characterized by katG463 CGG (Arg) and gyrA95 ACC (Thr).
The red arrow indicates that strains belong to Group 3, characterized by katG463 CGG (Arg) and gyrA95 AGC (Ser) (Brosch et al. 2002)8.4.2 Spoligotyping
Spacer oligotyping (spoligotyping) is a rapid, PCR-based method for identifying and genotyping members of the MTC. It is a robust technique, and the data are repeatable and easily exchangeable between laboratories making it the most widely used of the typing techniques. Its usage has been adopted globally because the data are presented in a binary digital form, and it is the largest current collection of the molecular characteristic of M. bovis (Driscoll 2009). This method exploits 31-41- bp-long, nonrepetitive spacers interspersed within short repetitive units of clustered, regulatory, short, palindromic repeats (CRISPR), referred to as the direct repeat (DR) region (Van der Zanden et al. 2002). Although there are more than 90 spacers in the DR unit of the MTC, the universally accepted panel contains only 43 spacers (Kamerbeek et al. 1997; He et al. 2012) that are immobilized by hybridization onto a membrane by southern blotting that produces a characteristic binary pattern depending on the presence or absence of each of the 43 spacers. Identification of members of the MTC (M. tuberculosis, M. bovis BCG, M. bovis, M. canettii, M. microti, M. africanum, and M. bovis subsp. caprae) is based on whether a sample contains the DR unit, in which case, there is a positive result on amplification. Genotyping is based on the different patterns produced by the presence or absence of the spacers. For example, the presence of all the spacers, except spacers 3, 9, 16, and 39 through 43, characterizes M. bovis. The resulting patterns are compared to those stored in the global spoligotype database (www.mbovis.org), and if unique, identity numbers are awarded to the new spoligotype patterns.
8.4.3 Interstitial Repetitive Unit-Variable Number of Tandem Repeat (MIRU-VNTR) Analysis
MIRU-VNTR is also a PCR-based typing tool that detects the number of repeats or alleles (Fig.
8.5) at each of the micro-repetitive satellites (loci) found on the mycobacterial genome (Allix et al. 2004). The number of repeats at each locus is determined by the weight of the PCR product for that particular locus and is designated by a number digit. The number of repeats at each of the loci chosen in a panel is a unique signature (expressed as integers) that is used for typing purposes (Fig. 8.6). The use of two panels (number of loci), the 15 and 24 loci panels, is recommended (van Soolingen et al. 1994; Frothingham and Meeker-O’Connell 1998; Roring et al. 2002; Supply et al. 2006). The choice of panel is dependent on cost, the required discriminatory power, and the polymorphism of the samples, which is usually a function of the geographical areas in question. Because of the likelihood of convergent evolution (homoplasy), it is advisable not to use this technique as a stand-alone molecular tool.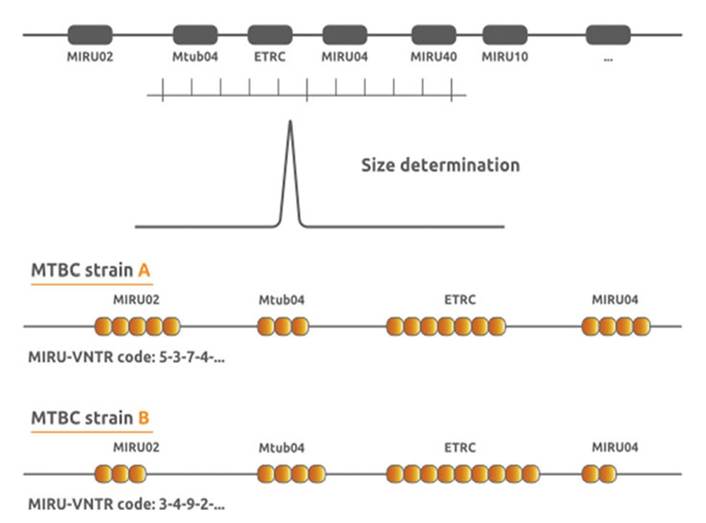
Fig. 8.5 MIRU-VNTR: The schematic representation of how repeats at micro-satellites (MIRU) are deduced
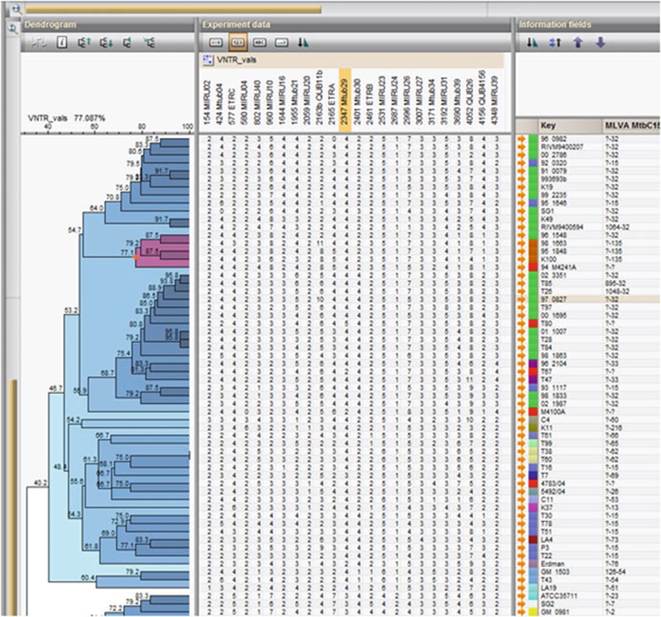
Fig. 8.6 An example of a typical MIRU-VNTR database showing how this tool is used for typing and phylogenetic purposes
8.4.2 Restriction Fragment Length Polymorphism (RFLP)
Restriction fragment length polymorphism (RFLP) is a partial genomic analysis technique that identifies DNA sequence polymorphism characterized by specific, nontandem, repeat units within specific regions of the mycobacterial genome (Durr et al. 2000). The detection of RFLPs is based on restriction enzyme digestion, and fragmentation of mycobacterial DNA in a genomic region where a specific short sequence is known to occur, followed by physical fragment separation by length using agarose gel electrophoresis (Fig. 8.7). A fragment length profile is considered an allele, and each one varies with an individual isolate thus forming the base for identification (Lin et al.
2014).Insertion sequence 6110 (IS6110) is the most extensively used genomic region (molecular marker) for epidemiological studies of M. tuberculosis including outbreak investigation, nosocomial infection, and multi-drug resistance studies (van Embden et al. 1992). Because of the presence of a relatively higher copy numbers
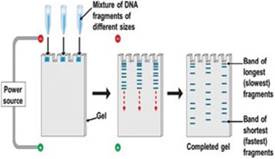
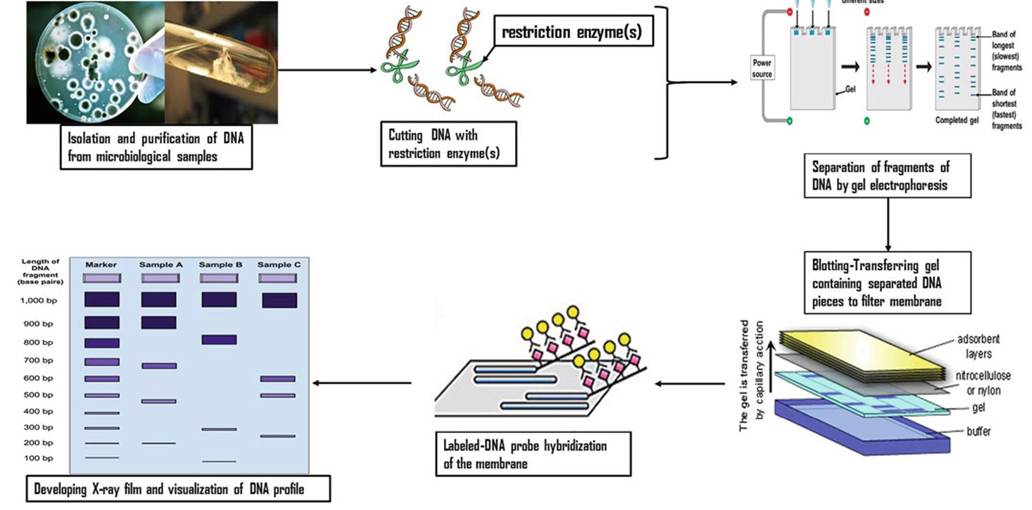
Fig. 8.7 Flow chart showing the steps involved in RFLP typing. This figure is an adaptation based on Komal (2014)
138 A. Muwonge et al.
(8-15) of the insertion sequence in the M. tuberculosis genome, RFLP analysis has a higher discriminatory power. By comparison, the majority of M. bovis isolates have fewer copies of IS6110 in their genomic structure (Kanduma et al. 2003). To compensate for the low discriminatory power of IS6110 because of the low copy number of insertion sequences, researchers proposed the use of the polymorphic GC-rich repeat sequence (PGRS) technique, as it is useful to differentiate between strains with fewer than six copies of IS6110 (van Embden et al. 1993; Yang et al.
2000). This PGRS-based RFLP probe is the most discriminatory of the probes currently available for M. bovis strain typing (Seva et al. 2014). Another novel RFLP probe, the pUCD probe that produces a highly polymorphic yet simple-to- analyze banding pattern, is also an effective tool for molecular typing of M. bovis strains (O’Brien et al. 2000; Cameron et al. 2001).
8.5
More on the topic Molecular Typing for Epidemiologic Studies of Mycobacterium bovis:
- Chapter 8 Molecular Epidemiology of Mycobacterium bovis in Africa
- TUBERCULOSIS, MYCOBACTERIUM BOVIS AND MYCOBACTERIUM CAPRAE INFECTIONS
- Mycobacterium bovis Infection in Humans
- The Use of Molecular Epidemiology in Understanding the Dynamics of M. bovis
- Molecular Epidemiology of M. bovis in Humans and Other Hosts in Africa
- Molecular Genetic Studies
- 5.3 Maintenance Hosts of Mycobacterium bovis
- Molecular Genetics of Mycobacterium avium subsp. paratuberculosis
- Genetic Diversity of Mycobacterium bovis Strains in Tanzania
- Mycobacterium bovis Infection in Humans in Egypt
- Chapter 4 The Control of Mycobacterium bovis Infections in Africa: A One Health Approach
- Chapter 22 Holes and Patches: An Account of Tuberculosis Caused by Mycobacterium bovis in Uganda
- Bacterial Culture and Typing Procedures
- Epidemiologic Problem Oriented Approach (EPOA) Methodology
- Molecular Epidemiology of Bovine Tuberculosis in Uganda
- Inferences on the Origin of M. bovis
- Molecular Epidemiology of BTB in Nigeria
- Human TB Due to M. bovis
- Public Heath Implication of M. bovis