Tumor mass heterogeneity
Based on the malignant transformation paradigm, the origin of cancer arises from a single initiated cell and corresponds with the accepted clonal nature of cancer development (Wainscoat & Fey, 1990).
However, during the processes of tumor promotion and progression, individual tumor cells can show distinct properties including differences in gene expression profiles, cellular morphologies, and biologic behaviors, which generate intratumoral heterogeneity. The development of tumor heterogeneity has the potential to compromise the successful treatment of cancer, not only in the context of tumor cell susceptibilities to anticancer therapeutics, but also the development of tumor cells with augmented invasive and metastatic capacities. Two main theories have been proposed to explain the development of tumor mass heterogeneity: (1) the clonal selection theory and (2) the cancer stem cell theory; both provide a conceptual framework for understanding the mechanisms of cancer progression.Clonal selection theory
The concept for the clonal evolution of cancer was first proposed in the late 20th century and sought to explain the observation that tumors often become more aggressive in their biologic behaviors, such as growth rate, invasiveness, and metastatic capacity, as a function of time (Greaves & Maley, 2012; Nowell, 1986). Clonal evolution of individual cancer cells within a tumor mass represents the sequential selection of variants derived from a common malignantly transformed clone. In this Darwinian model, driver mechanisms for clonal evolution of cancer cells are believed to be from the combined effects of internal and external factors. Intrinsically, the genetic instability of tumor cells, known as a mutator phenotype, serves as a principal driver for clonal evolution. On repeated rounds of initiated cell proliferation, the probability of acquiring additional genetic mutations continually increases, and a small fraction of mutated cells will have the chance to acquire some additional growth advantages.
Consequently, mutants with superior ‘fitness’ become the predominant subpopulations within a tumor mass. The co-existence of multiple ‘fit’ subpopulations provides the basis for tumor mass heterogeneity (Figure 3.4). In addition to the intrinsic genetic instability of tumor cells that contributes to clonal evolution, host factors that are operative within the tumor microenvironment are expected to exert selective pressures that drive the expansion of malignant clonal subpopulations. Host-derived factors involved in clonal evolution within the immediate tumor environment include immune surveillance, growth factors, and inflammation. 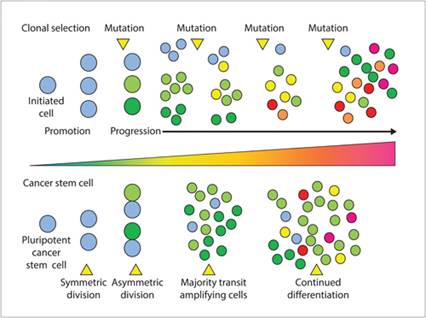
Figure 3.4 Different models for generating tumor heterogeneity. The clonal selection model suggests that genomic instability is the underlying mechanism for generating tumor heterogeneity. Genetic mutations that provide growth advantages will have the potential to drive the development of phenotypically different clonal subpopulations. The cancer stem cell model proposes that tumor heterogeneity is a consequence of a small number of pluripotent cancer stem cells capable of asymmetric division. Cancer stem cells give rise to transit amplifying populations, which are variably differentiated cancer cells with limited replicative potential.
Cancer stem cell theory
Stem cells are pluripotent cells with the capacity for indefinite proliferation and differentiation, and they serve as an inexhaustible cell source for replenishing normal cell populations undergoing homeostatic programmed cell death. Cancer stem cells are tumor cells that possess stem cell properties. The stem cell theory of cancer proposes that tumor mass heterogeneity is driven by a small fraction of malignantly transformed stem cells with the capacity to (1) divide and expand the cancer stem cell pool and (2) differentiate into heterogeneous non-tumorigenic cancer cell types that constitute the bulk of cells within a tumor mass (Figure 3.4).
Cancer stem cell division can be asymmetric, with daughter cell progenies being either stem cell in nature or not. Non-cancer stem cell progeny are categorized as transit amplifying cells. The specific population of transit amplifying cells demonstrates limited replication potential yet constitutes the major proportion of cells within a tumor mass. In contrast with transit amplifying cells, cancer stem cells comprise only a small percentage of the tumor cell mass and generally remain divisionally silent for the majority of the time. Definitive evidence supporting the cancer stem cell theory was derived from studies of acute myelogenous leukemia, where a small population of CD34+CD38– cells were capable of tumorigenesis and recapitulating tumor cell heterogeneity in a NOD/SCID murine preclinical model of leukemia (Bonnet & Dick, 1997). Shortly following the identification of cancer stem cells in hematologic malignancies, evidence for their existence in solid tumor histologies was first described in human glioma (Ignatova et al., 2002). To date, ample scientific evidence has been gathered to support the existence of cancer stem cells across a diverse group of both hematopoietic and solid tumor histologies (Prager et al., 2019).
The implications for the cancer stem cell theory are significant, as it indicates that only a small fraction of cancer cells within a tumor mass are fundamentally responsible for the genesis, maintenance, and recurrence of cancer. As such, thousands of scientific investigations have focused on understanding the properties of cancer stem cells with the intent of identifying the cellular behaviors and vulnerabilities that could be exploited for improving cancer prevention and treatment (Ju et al., 2022).
Hallmarks of cancer
Given the genetic basis of cancer and the diverse mutagenic stimuli that a vast number of germline and somatic cells are exposed to routinely, it is remarkable that the generation of renegade cells and consequent development of macroscopic tumor masses are relatively infrequent events in complex, multicellular organisms.
Largely by virtue of the inherent genetic safeguards such as cell cycle arrest, programmed cell death, and DNA repair mechanisms, host organisms survive for an entire lifetime without fatal cancer development. Nonetheless, in rare instances where all steps of malignant transformation are achieved, cells display conserved aberrant biologic behaviors that serve as foundational abnormalities shared by cancer cells. This constellation of deranged cellular activities and properties are considered the seminal hallmarks of cancer (Hanahan & Weinberg, 2000, 2011).Self-sufficiency in growth signals
Normal cell growth and proliferation require the transcriptional activation of genes responsible for cell cycling. Such stimulatory intracellular signals can be derived from the binding of cell surface receptors with diffusible growth factors, extracellular matrix components, or cell-to-cell interactions. In the absence of external stimulation, normal cells will not proliferate, but rather remain quiescent in the G0 phase. In contrast, cancer cells acquire genetic mutations that do not have the requirement of exogenous stimulatory signals for cell replication and are deemed growth self-sufficient. The achievement of growth self-sufficiency can be accomplished by cancer cells through different genetic alterations that are most commonly are associated with oncogene activation. Classical oncogenic mutations leading to growth self-sufficiency include excessive growth factor production, overexpression of growth receptors, constitutive activation of signaling pathways, and disruption of negative feedback mechanisms responsible for terminating proliferative responses.
Insensitivity to antigrowth signals
Complementing the actions of growth self-sufficiency, cancer cells also exhibit insensitivity to antigrowth signals, and in combination, both properties promote and accelerate the unrestrained proliferation of cancer cells. Normal cells will obey signals derived from the immediate microenvironment, including cues provided by neighboring cells and extracellular matrix, to arrest from the cell cycle and terminally differentiate.
However, genetic mutations in tumor suppressor genes, such as P53 and RB, can endow cancer cells with insensitivities to antigrowth signaling, with consequent unrestricted cell cycling and proliferation.Evading apoptosis
Apoptosis is a form of programmed cell death and serves as a homeostatic mechanism to allow normal cells to be removed from the host organism. The apoptotic program involves hundreds of proteins; however, p53 serves as a master regulator. As such, mutations in the P53 gene can endow cancer cells with the ability to evade programmed cell death, even in the face of catastrophic genomic injury. Blocks in the apoptotic program in cancer cells are common and can be mediated through multiple mechanisms including downregulation of membrane death receptors, increased anti-apoptotic protein expression, and sequestration of initiator and executioner caspases.
Limitless replicative potential
Normal cells have a finite replicative potential, which is dictated by the rate of successive telomere length erosion following each round of DNA replication. Except for normal pluripotent stem cells, normal somatic cells following repeated cellular divisions will ultimately undergo a process called senescence and lose the capacity for further replication. Mechanistically, finite replication due to telomere erosion serves as a protective barrier to prevent highly mutagenic processes such as breakage–fusion–bridge cycling, which is the inadvertent fusion of sister chromatid pairs during mitosis. Cancer cells express telomerase, an enzyme typically used by pluripotent stem cells that allows for the maintenance of telomere length with consequent endowment of infinite replicative potential. The use of telomerase or other telomere lengthening strategies by cancer cells serves as a conserved mechanism allowing the achievement of cellular immortalization.
Sustained angiogenesis
Angiogenesis is the process by which new blood vessels are derived from pre-existing vasculature.
Based on the limits of nutrient and oxygen diffusion, macroscopic tissue growth exceeding 1–2 mm in diameter requires the establishment of new blood vessels. The regulation of angiogenesis is complex and requires the coordinated interplay and balance between pro- and anti-angiogenic peptides that act on endothelial cells. Under normal physiological conditions, angiogenesis can be transient and reversible, as demonstrated in normal wound healing. However, cancer cells favor activation of the angiogenic switch, which shifts the balance to sustained new blood vessel formation and thereby allows for the continued macroscopic growth of tumor cell masses.Tissue invasion and metastasis
The outgrowth of cancer can be biologically categorized as being benign or malignant and is dependent on behavior attributes of cancer cell populations. The most problematic tumor histologies are those that invade and involve distant organs through the process of metastasis. Tissue invasion is mediated by the increased directional motility of cancer cells in conjunction with the liberation of proteases capable of degrading the basement membrane and associated extracellular matrix proteins including collagen, fibronectin, and gelatin. Tumor cell metastasis is the progressive extension of localized tissue invasion, where individual or small numbers of tumor cells have entered the circulatory system, either a blood or lymphatic vessel, and proceeded to disseminate to distant organs. The specific steps of this metastatic cascade can be categorized into discrete processes: (1) detachment from the primary tumor, (2) migration and intravasation, (3) circulatory transport, (4) arrest and extravasation, (5) resistance to anoikis and colonization, and (6) angiogenesis.
Two emerging hallmarks of cancer
Since the first description of the hallmarks of cancer in 2000 (Hanahan & Weinberg, 2000), new investigations conducted over the last decade have suggested that two additional characteristics reflective of tumor behavior should be considered as emerging hallmarks. Specifically, cancer cells appear to universally demonstrate properties that allow for the reprogramming of energy metabolism and evading immune destruction (Hanahan & Weinberg, 2011). With regards to altered energy metabolism, the capacity of cancer cells to preferentially utilize aerobic glycolysis has been recognized for close to 100 years, and is termed the Warburg effect. The underlying mechanism for cancer cells to preferentially utilize glycolysis as a main energy source remains enigmatic; however, it has been hypothesized that increased glycolysis might allow for the diversion of glycolytic intermediates into biosynthetic pathways that support rapid cellular proliferation, and hence provide a growth advantage for cancer cells (Vander Heiden et al., 2009). Addressing the capacity of tumor cells to evade immune destruction, substantive preclinical and epidemiologic evidence supports the important role of the immune system as a barrier to tumor formation and progression. As such, the presence of malignantly transformed cells, which progress to develop into macroscopic tumor burdens in immunocompetent hosts, would suggest that a conserved set of mechanisms are employed by cancer cells for immune system evasion.