Malignant transformation: a multistep process
With the identification that carcinogens were responsible for the development of cancer, scientists were able to study the key cellular events leading to overt tumor formation. It became evident, particularly in the study of chemical carcinogenesis, that the maladaptation to a cancerous phenotype required an individual cell to acquire several concurrent genetic or epigenetic perturbations, and such ‘steps’ towards a malignant phenotype could be categorized into three distinct stages: initiation, promotion, and progression (Boyland, 1985; Foulds, 1954, 1965).
The ultimate outcome of this multistep process is the development of cancer cells with invasive properties (Figure 3.3). 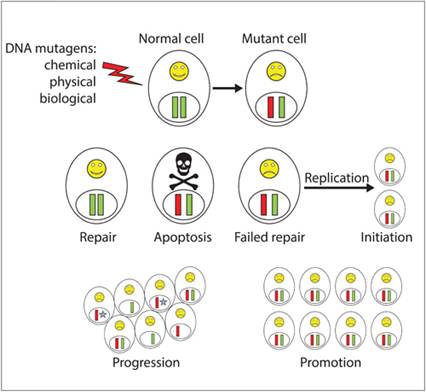
Figure 3.3 Steps involved in cellular malignant transformation. Following a mutagenic event, normal cellular outcomes include cell cycle arrest with complete DNA repair, catastrophic and irreparable DNA damage leading to apoptosis, or failed DNA repair. Perpetuation of heritable genomic defects to daughter cells represents the initiation phase of malignant transformation. Clonal expansion of initiated cells and acquisition of additional genetic mutations represent the steps of tumor promotion and progression, respectively.
Initiation
The potential for tumor initiation starts with any mutagenic event leading to a change in cellular DNA, often a single base alteration, in susceptible cells. Following the induction of DNA mutation, cellular outcomes include (1) programmed cell death, (2) cell cycle arrest with repair of DNA damage, or (3) failure to repair DNA damage. In cells undergoing apoptosis or successfully repairing DNA damage, the mutagenic event and potential for carcinogenesis are completely neutralized. However, in cells failing to repair DNA mutations and subsequent procession through DNA replication, the production of heritable genome changes becomes irreversible and constitutes the event of ‘initiation’.
However, not all initiated cells will proceed forward to establish cancer, as some initiated cells will harbor silent genetic mutations given that 98.5% of the genome is not responsible for protein coding. Additionally, the cellular outcomes of initiated cells can be dormancy or apoptosis. As such, an initiated cell is not synonymous with a tumor cell, as the genomic alterations of an initiated cell might remain undetectable throughout the life of the host organism unless additional genomic perturbations are acquired that further promote cell proliferation and genomic instability.Promotion
During tumor promotion, initiated cells are provided with a selective growth advantage through transient increases in cell division, decrease in apoptosis, or a combination (Wright et al., 1994). Clonal expansion of initiated cell populations can occur following exposure to non-genotoxic or weakly genotoxic agents and result in the formation of pre-malignant lesions (Slaga, 1983). Many chemicals have been identified that act as tumor promoters, with croton oil, tetradecanoyl phorbol acetate, and phenobarbital serving as examples. In addition to chemical agents, tumor promotion can be elicited by trauma or cell death, which results in inflammation and the release of growth, survival, and pro-angiogenic factors (Grivennikov & Karin, 2010; Rundhaug & Fischer, 2010). Real life examples that might be striking are the habit of drinking alcohol and smoking as a pastime, with combustion of cigarette smoke providing the carcinogens, and drinking alcohol serving as a promoting agent through the induction of gingival mucosal cell death and compensatory cell proliferation (Pelucchi et al., 2006).
Progression
During the clonal expansion of initiated cell populations, pre-malignant cells have the potential to acquire additional genetic mutations, and a major hallmark of tumor progression is chromosomal instability (Loeb, 1991; Nowell, 1976). As a result of genetic instability, pre-malignant cells acquire additional karyotype alterations that allow for increased growth speed, invasiveness, and metastatic potential.
Many of the chromosomal abnormalities that accumulate during tumor progression include mutations that inactivate tumor suppressor genes and activate oncogenes.Tumor suppressor genes
Genes classified as tumor suppressors have the capacity to protect normal cells from malignant transformation. Classically, tumor suppressor genes have been described as acting recessively and as following the ‘two-hit hypothesis’, originally proposed by Alfred G. Knudson in 1971 (Knudson, 1971), which implies that both maternal and paternal alleles that encode a specific tumor suppressor gene must be affected before a deficient phenotype is produced. Despite the validity of the ‘two-hit hypothesis’ for many genes, altered phenotypes can be produced because of genetic change within one parental allele; such exceptions include haploinsufficiency and dominant negative mutations (Payne & Kemp, 2005), whereby the phenotype is blended and not dichotomous. Broadly, tumor suppressor genes can be categorized as ‘gatekeepers’ or ‘caregivers’, and more recently ‘landscapers’ based on their different mechanisms for minimizing heritable genomic changes (Deininger, 1999; Levitt & Hickson, 2002).
Gatekeepers
‘Gatekeeper’ genes function by directly controlling cell growth via cell cycle regulation or promote programmed cell death. Gatekeeper genes are principally responsible for protecting against tumor cell initiation, which is the first and critical step in the malignant transformation process. Mutations in gatekeeper genes can occur at both the somatic and germline levels, with sporadic tumors more frequently having somatic mutations and hereditary tumor syndromes driven by germline mutations in gatekeeper genes (Vogelstein & Kinzler, 2004). Li-Fraumeni syndrome and Retinoblastoma are examples of hereditary tumor syndromes in which germline mutations occur in P53 and retinoblastoma (RB) genes, respectively (Strahm & Malkin, 2006).
The two most notable gatekeeper tumor suppressor genes are the P53 gene and the RB gene, both serving critical functions in regulating the cell cycle (Sager, 1992).
Referred to as the ‘Guardian’, of the genome, the p53 protein is responsible for initiating multiple cellular programs that prevent malignant transformation, including the activation of DNA repair proteins, arrestment of the cell cycle at the G1/S checkpoint, and induction of apoptosis (Efeyan & Serrano, 2007). In parallel with the P53 gene, the RB gene serves as a master regulator of cell cycle progression in the G1 phase. Early in the G1 phase, Rb protein exists in a hypophosphorylated state and binds tightly with E2F family transcription factors (Cobrinik et al., 1992). The association of Rb protein with E2F family transcription factors prevents the transcription of target genes required to progress past the G1/S checkpoint. Upon progressive phosphorylation of the Rb protein by active CDKs, E2F family transcription factors are released by the Rb protein, allowing cells to progress into the DNA synthesis phase of the cell cycle (Cobrinik et al., 1992; Hamel et al., 1992). Since the discovery of the P53 and RB genes as tumor suppressor genes, a multitude of additional gatekeeper tumor suppressor genes have been identified (Table 3.2).Table 3.2 Abbreviated list of recognized tumor suppressor genes
| Major caregiver genes | ||
| Gene | Protein function | Associated tumors |
| APC | Cell adhesion, signal transduction pathway | Colorectal cancer |
| VHL | Transcriptional elongation regulation | Schwannoma, meningioma, others |
| PTEN | Phosphatase | Hamartoma, glioma, others |
| RB1 | Cell cycle control | Osteosarcoma, others |
| TP53 | Cell cycle control, apoptosis | Sarcoma, leukemia, others |
| NF1 | Ras GAP activity | Neurofibroma, sarcoma, others |
| CDKN2A | Cell cycle control | Melanoma, pancreatic cancer |
| WT1 | Transcription factor | Nephroblastoma |
| BRCA1 | DNA repair, cycle checkpoint control | Breast and ovarian cancer |
| BRCA2 | DNA repair, cycle checkpoint control | Breast and ovarian cancer |
| ATM | DNA repair | Lymphoma |
| FANCA | DNA repair | Acute myeloid leukemia |
| MLH1 | DNA mismatch repair | Lymphoma, sarcoma, others |
| NER | Nucleotide excision repair | Skin cancer |
Caregivers
‘Caregiver’ genes are another category of genes responsible for protecting the genome. These genes are involved in maintaining genomic stability, principally through regulation of DNA repair pathways, which reduces the mutational rate of the host genome (Goode et al., 2002).
Unlike gatekeeper genes, which are principally involved in protecting against tumor cell initiation, caregiver genes play a larger role in the tumor progression stage of malignant transformation. Mutations in caregiver genes have the potential to accelerate the multistep tumorigenic process simply because of enhanced genomic instability and consequent acquisition of additional genetic mutations. The importance of caregiver genes is highlighted by the increased likelihood for developing hereditary breast and ovarian cancer in women harboring BRCA1 and BRCA2 gene mutations (King et al., 2003; Szabo & King, 1995). Mechanistically, BRCA1 and BRCA2 proteins participate in the formation of a large multi-subunit protein complex called BRCA1-associated genome surveillance complex, which is critical for the identification and repair of double-strand DNA breaks through homologous recombination (Liu & West, 2002). Ineffective repair of DNA breaks, in particular double-strand breaks, dramatically increases the risk for progressive genomic instability and cancer development (Karran, 2000; Moynahan et al., 1999). In human cancer patients with BRCA1 and BRCA2 mutations, inhibitors of PARP have proved to be exceptionally effective in killing mutated cancer cells through a process known as ‘synthetic lethality’ (Helleday, 2011). Although not as extensive as gatekeeper genes, many tumor suppressor genes that function as caregivers and participate in cancer susceptibility have been identified and characterized (Table 3.2).Landscaper genes
In addition to gatekeeper and caregiver tumor suppressor genes, a third category of genes with suppressive properties has been proposed and termed ‘landscaper’ genes. As suggested by their name, landscaper genes act on the microenvironment in which cells reside (Cooper & Giancotti, 2019). Interestingly, it is well accepted that cellular behaviors can be directly affected by the microenvironment, a process known as ‘outside to inside’ signaling, often mediated by integrins.
Aberrations in landscaper genes that are responsible for mechanical properties of the extracellular matrix could result in changes in cellular behaviors, as well as alter microenvironmental cues including growth factors, cell adhesion molecules, and extracellular matrix properties, which in turn can influence the behavior of resident-initiated cells (Kinzler & Vogelstein, 1998).Oncogenes
Proto-oncogenes code for normal cellular machinery involved in cell growth and differentiation, which includes growth factors, growth factor receptors, protein kinases, adaptor proteins, G-protein signaling transducers, and transcription factors (Table 3.3). Genetic alterations capable of dysregulating the expression or activity of proto-oncogenes can lead to dominant, gain-of-function mutations with the consequent generation of an active oncogene. Unlike tumor suppressor genes, conversion of a proto-oncogene to an oncogene necessitates only one parental allele to be transformed within a susceptible cell to obtain a phenotypic change. The conversion of proto-oncogenes to hyperactive oncogenes can be categorized into at least four mutagenic mechanisms: (1) point mutations, (2) gene amplification, (3) chromosomal translocation, and (4) viral insertions.
Table 3.3 Abbreviated list of recognized oncogenes
| Gene | Protein function | Associated tumors |
| ALK | Receptor tyrosine kinase | Lymphoma |
| BCL-2 | Anti-apoptotic protein | Lymphoma, leukemia |
| C-MYC | Transcription factor | Leukemia, carcinoma, others |
| EGFR | Cell surface receptor | Squamous cell carcinoma |
| GLI | Transcription factor | Glioblastoma |
| KIT | Receptor tyrosine kinase | Sarcoma, gastrointestinal stromal tumor, others |
| JUN | Transcription factor | Sarcoma |
| RAS | G-protein signal transduction | Carcinoma |
| RET | Receptor tyrosine kinase | Thyroid carcinoma, multiple endocrine neoplasia |
| SIS | Growth factor | Glioma, fibrosarcoma |
| SRC | Tyrosine kinase | Sarcoma |
| TRK | Receptor tyrosine kinase | Colon and thyroid carcinoma |
Point mutations can produce phenotypic changes either through the generation of proteins resistant to normal regulatory cues or through degradation pathways, resulting in constitutive activation or functional hyperactivation. The RAS oncogene transcribes a protein harboring a point mutation, which endows resistance to normal regulatory enzymatic activity (GTPase activity) and consequently allows for sustained and dysregulated intracellular signaling (Hamilton & Vogelstein, 1988).
The identification of homogeneously staining regions and double minutes are genetic hallmarks of gene amplification, which can result in the transformation of a proto-oncogene to an oncogene. The translational product of gene amplification is normal; however, the absolute quantities of protein can be log orders greater than normal given the dramatic increases in mRNA transcripts. The overexpression of human epidermal growth factor receptor 2 (HER2) in aggressive breast cancer is an example of gene amplification that serves as a drug target for improving cancer management using an antibody called Herceptin (Ross & Fletcher, 1999).
Chromosome translocations can result from the joining of different chromosome arms and have the potential to produce excessive levels of normal or novel proteins because of coupling strong promoter sequences upstream of proto-oncogene coding regions. As such, chromosomal translocation serves as one genetic mechanism responsible for the conversion of proto-oncogenes to oncogenes. Perhaps the most well studied and therapeutically targetable chromosomal translocation is the Philadelphia chromosome, which is a balanced chromosomal translocation between chromosomes 9 and 22 (Koretzky, 2007). The Philadelphia chromosome produces a novel BCR–ABL fusion gene capable of producing a protein with excessive tyrosine kinase activities and serves as the oncogenic mutation responsible for the development of chronic myeloid leukemia (Konopka & Witte, 1985; Westbrook, 1988). The drug Gleevec was engineered to inhibit the BCR–ABL fusion protein and is first-line therapy for human cancer patients diagnosed with chronic myelogenous leukemia (Iqbal & Iqbal, 2014).
Historically, the discovery and characterization of oncogenes were first described in studies of viruses with cancer forming properties. Mechanistically, retroviruses exert their oncogenic effects through a process termed insertional mutagenesis, whereby viral genetic material is inserted into the host cell’s genome. Differences in penetrance and latency of tumor development following retroviral infection are associated with the type of retrovirus and classified as either acute or late transforming. Acute transforming retroviruses carry viral oncogenes (v-onc) within their genome, and on infection of host cells, the transcription of v-onc is driven by strong viral promoter sequences found within the 5′ long terminal repeat sequences. Consequently, malignant transformation occurs rapidly following infection with acute transforming retroviruses (Gray, 1991; Uren et al., 2005). In contrast, late transforming retroviruses do not carry viral oncogenes within their genome, and therefore are not likely to induce rapid malignant transformation. Rather, late transforming retroviruses randomly insert into the host cell genome with the low incident possibility of being inserted in proximity to a normal cellular proto-oncogene. In these rare instances, strong viral promoters and enhancers of late transforming retroviruses are capable of hijacking the transcription of proximal cellular proto-oncogenes, resulting in gain-of-function activities (Gray, 1991; Uren et al., 2005). Retroviral infection as a cause of malignancy in human beings is rare, with one example being human T-cell lymphotropic virus type 1, which is associated with the development of adult T-cell leukemia/lymphoma (Robert-Guroff et al., 1985). In parallel, several retroviruses are responsible for the development of cancers in felines and include feline leukemia virus, feline immunodeficiency virus, and feline sarcoma virus (Beatty, 2014; Fujinaga & Green, 1971; Hoover & Mullins, 1991; Jarrett, 1975; Linenberger & Abkowitz, 1995; McDonough et al., 1971).
More on the topic Malignant transformation: a multistep process:
- Malignant transformation: a multistep process
- Barger A.M., MacNeill A.L. (Eds.). Small Animal Cytologic Diagnosis: Canine and Feline Disease. CRC Press,2024. — 536 p., 2024
- Small intestine (duodenum, jejunum, and ileum)