COOPERATION OF DOMINANT TRANSCRIPTION FACTORS
In the same co-culture model of cells expressing the HIV-1 Env with cells expressing CD4, NF-B, p53, and AP1 are activated in response to the initiation of Env-elicited apoptosis.9 The repressor
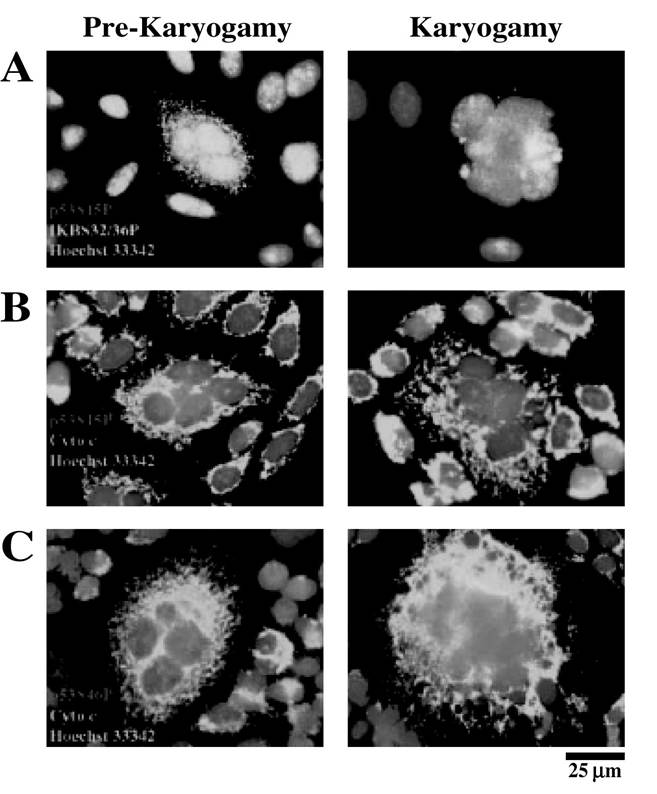
FIGURE 16.2 p53 phosphorylation in Env-dependent syncytia.
(A-C) Representative immunofluorescence microphotographs of prekaryogamic (18 h) and karyogamic syncytia (36 h) labeled with antibodies specific for I-B S32/36P (A), p53S15P (A and B, light area surrounding dark area), p53S46P (A and C, dark dots in the circumference), or cytochrome c (B and C, light area). Nuclei are stained with Hoechst 33342 for chromatin condensation.NF-B exhibits antimitotic and antiapoptotic effects and prevents the Env-elicited phosphorylation of p53 on serine 15 and 46, as well as the activation of AP1. Thus, NF-B inhibition fully prevents the syncytial accumulation of cyclin B1, the p53-dependent transcription and phosphorylation, and karyogamy.9 Therefore, NF-B exerts its action on the aberrant cyclin B1/Cdk1-mediated entry into mitosis and upstream p53 activation.
Transfection with dominant-negative p53 abolished syncytial apoptosis and AP1 activation and increased the lifetime of syncytia.9 In addition, 85% of the transcriptional effects of HIV-1 Env were blocked by the p53 pharmacologic inhibitor pifithrin-, as revealed by microarrays.9 Moreover, macroarrays identified Puma, a proapoptotic “BH3-only” protein from the Bcl-2 family known to activate Bax/Bak,14 as being transcribed in response to Env, in a p53-dependent manner (Figure 16.1). Downmodulation of Puma by antisense oligonucleotides, as well as RNA interference of Bax and
Bak, prevented Env-induced apoptosis. HIV-1-infected primary lymphoblasts upregulated Puma in vitro.f9 Accordingly, circulating CD4+ lymphocytes from untreated HIV-1-infected donors contained enhanced amounts of Puma protein, and these elevated Puma levels dropped with administration of antiretroviral therapy.
NF-B and p53 activation were observed in lymph node biopsies of HIV-1- infected patients.9 Altogether, these results establish that NF-B and p53 are the dominant and competing transcription factors participating in HIV-1 infection, CD4+ depletion, and syncytial death.Mitochondrial and caspase activation
Depending on the model studied (e.g., the cell type, the apoptotic signal), apoptosis can be promoted via two general pathways: the death receptor pathway (e.g., Fas/CD95, TNF) or the mitochondrial pathway, namely, the intrinsic pathway (Figure 16.3). These pathways occur preferentially in type-I cells and in type-II cells,15 and also in HIV-1-mediated cell death.16 The mitochondrial pathway is characterized by a mitochondrial checkpoint (for review see Kroemer and Reed17), corresponding to an increase in MMP and a subsequent relocalization of apoptogenic
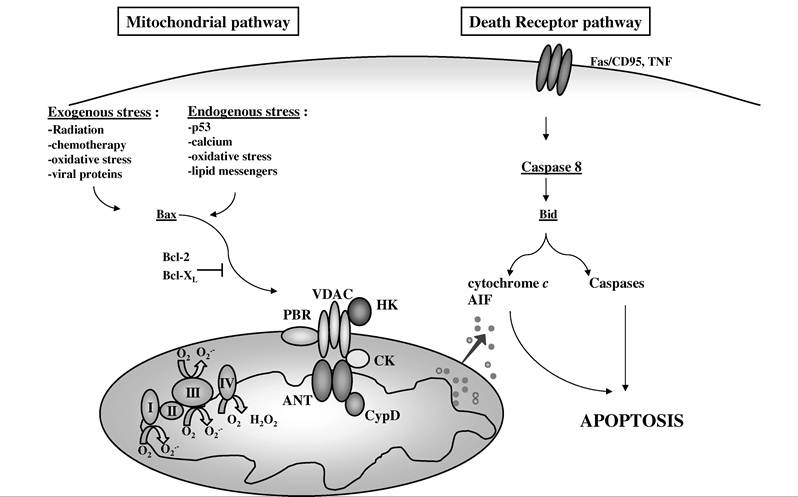
FIGURE 16.3 The signaling pathways of apoptosis. Apoptosis can be initiated via two major pathways: the death receptor pathway and the mitochondrial pathway. The occurrence of one pathway may be dependent on the cell type (type I vs. type II) or the type of proapoptotic trigger. Endogenous stress signal as well as endogenous stimuli that converge to the mitochondrion elicit the mitochondrial pathway. As a consequence, the mitochondrial membrane permeabilizes, intermembrane space proteins (e.g., cytochrome c, AIF) are released, and the degradation phase of apoptosis that leads to cell death is initiated. Constitutive mitochondrial proteins from the respiratory chain (complexes I, II, III, and IV, CI, CII, CIII, and CIV) and the permeability transition pore complex (peripheral benzodiazepine receptor [PBR], voltage-dependent anion channel [VDAC], hexokinase [HK], creatine kinase [CK], adenine nucleotide translocase [ANT], cyclophilin D [CypD]) can contribute to the process of membrane permeabilization that is regulated by the Bax/Bcl-2 family proteins.
In contrast, the death receptor pathway is triggered by exogenous ligands of the cell surface receptors, such as Fas/CD95 or TNF. The early activation of caspase-8 and the cleavage of the proapoptotic protein Bid characterize this pathway. In some cases, this pathway can also be inhibited by Bcl-2.proteins from the mitochondria into the cytosol, such as cytochrome c, AIF, second mitochondria- derived activator of caspases (Smac)/direct inhibitors of apoptosis protein-binding protein with low pI (DIABLO), procaspases, and Endo G.18-20 Although the precise mechanism of release of these various proteins is still unclear, they may cross the outer mitochondrial membrane via either specific or nonspecific pores. These pores are composed of oligomerized members from the Bax/Bcl-2 family (Bax, Bid)21 and may also contain other proteins, such as the voltage-dependent anion channel (VDAC)22 or the adenine nucleotide translocase (ANT), two members of the permeability transition pore complex (PTPC).23
As described above, as an intermediate stage of syncytial death, phosphorylation of p53 leads to an increased expression of Bax and its translocation to mitochondrial membranes, triggering the activation of mitochondria and caspases. Thus, after a conformational activation of Bax and the exposure of its N-terminus, the permeability of mitochondrial membranes is increased and allows for the release of intermembrane proteins such as cytochrome c and AIF from mitochondria.6 This phase is independent of caspases, because their inhibition does not suppress the translocation of AIF and cytochrome c, yet it prevents all signs of nuclear apoptosis. In contrast, translocation of Bax to mitochondria leads to MMP, which is inhibited by microinjected Bcl-2 protein or Bcl-2 gene transfection.6 Bcl-2 also prevents the subsequent nuclear chromatin condensation and DNA fragmentation. The two proteins initiate two different cascades of events downstream of MMP. Thus, microinjection of AIF into syncytia suffices to trigger rapid, caspase-independent cytochrome c release. Neutralization of endogenous AIF by injection of an antibody prevents all signs of spontaneous apoptosis occurring in syncytia, including the cytochrome c release and nuclear apoptosis. In contrast, cytochrome c neutralization prevents only nuclear apoptosis and does not affect AIF release.6
HIV-1 protease inhibitors can inhibit apoptosis at the mitochondrial level, which might help to prevent the HIV-1-mediated loss of lymphoid cells.24 Thus, it seems that the pathogenesis of AIDS and the pharmacologic interventions associated with this disease affect the mitochondrial regulation of apoptosis, which, therefore, largely determines the outcome of HIV-1 infection. In the future, it would be interesting to determine whether HIV-1 protease inhibitors can also inhibit syncytial death.