THE FORMATION OF SYNCYTIA
Syncytia are multinucleated giant cells resulting from a cell-to-cell fusion mechanism. Syncytia can be induced by the fusogenic action of cytokines (e.g., IL-4- and IL-13-induced fusion of monocytes-macrophages), as well as by receptor-ligand interactions at the cell surface (e.g., HIV- 1-infected T lymphocytes fusion, osteoclast activation during osteoporosis).
Thus, syncytia formation can be a physiological process and can contribute to disease progression by accelerating the cell death of infected cells, such as HIV-1-infected cells.10Among HIV-1 proteins, gp120 may be the most important apoptogenic protein. The glycoprotein (gp)120 is expressed in a complex with gp41, namely, the envelope glycoprotein complex Env, on HIV-1 virions and at the surface of HIV-1-infected cells. The gp120 interacts with CD4, a cell surface receptor, and CXCR4 or CCR5, two co-receptors belonging to the chemokine receptor family.4 The gp41 allows membrane interaction between a fusion domain of the protein and the cell surface. These Env-driven protein interactions trigger an apoptosis signal that activates a cascade of events leading to syncytia formation or single-cell killing.11 In syncytial death, the interaction of gp120 triggers the exposure of gp41 and favors insertion into the cellular membrane of a hydrophobic fusion peptide. In turn, the structure of gp41 is rearranged into a six-helix hairpin structure linking together cellular membranes and allowing them to fuse.12 After membrane fusion, death follows in a process involving deregulation of mitosis, karyogamy, caspase activation, inhibition of survival signals, and mitochondrial membrane permeabilization (MMP; for reviews see Castedo et al.5,13).
Deregulation of Mitosis and Karyogamy
Syncytia resulting from the fusion of cells expressing an Env gene with cells expressing the CD4/CXCR4 complex undergo apoptosis through a mitochondrion-controlled pathway that is initiated by the accumulation of cyclin B and the activation of cyclin B-dependent kinase 1 (CDK1; Figure 16.1).7,8 This step is followed by the phosphorylation of p53 on serine 15 by the mammalian target of rapamycin (mTOR/FRAP).
This activates p53-mediated transcription, which results in the expression of genes that induce MMP and cell death (Figure 16.1). This pathway depends on the interaction of Env with co-receptors, because pharmacologic inhibitors targeting gp41 and gp120 delay the formation and the death of syncytia. Moreover, pharmacologic inhibitors such as inhibitors of CDK1 (e.g., roscovitin), mTOR/FRAP (e.g., rapamycin), and p53 (e.g., cyclic pifithrin-α), or overexpression of Bcl-2, a mitochondrial oncoprotein known to inhibit MMP and apoptosis, all independently reduce the syncytial death (Figure 16.1).7,8In syncytia, CDK1 induces a phosphorylation/depolymerization of lamin that culminates in the process of the fusion of nuclei, i.e., karyogamy, before the entry of the cell into an early mitotic prophase (Figure 16.1).7,8 Thus, experimental manipulations that decrease CDK1 activity, such as microinjection of an antibody against CDK1, pharmacologic inhibition, and transient transfection of syncytia with a dominant-negative gene of CDK1, prevent the karyogamy. As a consequence, these procedures inhibit all the subsequent hallmarks of apoptosis.7,8
Subsequent to CDK1 activation, mTOR/FRAP that is located in the cytosol translocates to the nucleus and interacts physically with p53 to phosphorylate it on two serine residues (Figure 16.2).7,8 The relocation of mTOR/FRAP and the phosphorylation of p53 do not occur in roscovitin-treated syncytia, attesting to the importance of CDK1 in this process. Moreover, inhibition of mTOR/ FRAP by rapamycin, its specific pharmacologic inhibitor, reduces apoptosis in several paradigms of syncytium-dependent death, including primary CD4+ lymphoblasts infected by HIV-1. Concomitantly, rapamycin inhibits p53 phosphorylation on serine 15 (p53S15), mitochondrial translocation of Bax, loss of mitochondrial transmembrane potential (∆Ψm), mitochondrial release of apoptogenic proteins (e.g., cytochrome c and apoptosis-inducing factor [AIF]), and nuclear chromatin condensation.
In addition, transfection with the dominant negative p53 gene has a similar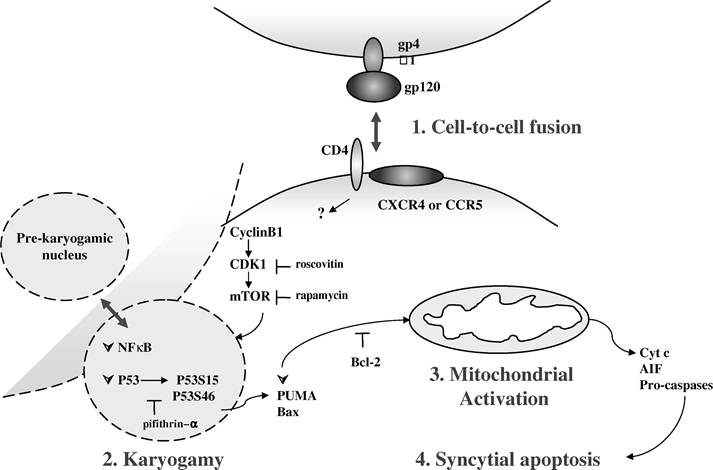
FIGURE 16.1 The syncytial death. Syncytia are multinucleated giant cells arising from the fusion of HIV-1 Env-expressing cells and CD4- and CCR5/CXCR4-expressing cells. After formation, syncytia die by apoptosis after a four-step process. (1) Cell-to-cell fusion: The interaction of cell surface proteins (gp120-GP41 complex or Env, CD4, CXCR4, or CCR5) promotes the fusion of membrane and cytoplasm, the accumulation of cyclin B1, and the activation of CDK1. (2) Karyogamy: This elicits the nucleoplasm fusion, the karyogamy, and the cooperation of two dominant transcription factors, p53, and NF-B. Thus, after nuclear translocation, the kinase mTOR/FRAP phosphorylates p53 on serine 15 and 46, which induces an increased expression of the proap- optotic Puma and Bax. (3) Mitochondrial activation: Mitochondria and caspase activations that manifest by membrane permeabilization, the release of proteins (e.g., cytochrome c and AIF), follow these events into the cytosol and, finally, the chromatin condensation, and (4) syncytial apoptosis. CDK1, mTOR/FRAP, and P53- specific pharmacologic agents as well as Bcl-2 can inhibit syncytial death.
antiapoptotic action as rapamycin, upstream of the Bax upregulation/translocation.7 Among a panel of kinases known to phosphorylate p53, including ERK, p38 kinase, DNA-PK, ATM, ATR, and mTOR/FRAP, only the mTOR/FRAP expression was enhanced during syncytia formation. The expression of the other kinases was unaffected. In contrast, the protein phosphatase PP2A that inhibits mTOR/FRAP was downregulated at a late stage of syncytia, well after mTOR/FRAP upregulation.7 Thus, the ability of mTOR/FRAP to initiate phosphorylation of p53 on serine 15 is associated with the initiation of syncytial death.7
Interestingly, the p53S15 phosphorylation and mTOR/FRAP upregulation occur also in vivo, in HIV-1-infected patients, in which it can be detected in pre-apoptotic and apoptotic syncytia in lymph nodes as well as in peripheral blood mononuclear cells, correlating with viral load.8 In summary, phosphorylation of p53S15 by mTOR/FRAP plays a critical role in syncytial apoptosis driven by HIV-1 Env.