HIV PR AND CELL DEATH
Evidence of Cell Death Caused by HIV PR
Necrosis
There is some evidence that expression of HIV-1 PR can cause cell death through necrosis. In yeast, the expression of HIV PR causes yeast growth arrest and cell lysis.7 The growth arrest of yeast-expressing HIV-1 PR occurs 14 to 18 h after induction, which is coincident with the peak of HIV-1 PR expression as detected by Western blotting.
The lytic activity of HIV-1 PR in yeast is evidenced by the release of intracellular protein into the culture medium from damaged cells. Ultrastructural examination by electron microscopy shows morphological alterations of the cells expressing HIV-1 PR including altered cell size, detachment of the plasma membrane from the cell wall, and a normal appearance of the nucleus. COS cells that express HIV PR through infection using a hybrid vaccinia virus-T7 RNA polymerase expression system exhibit a necrotic phenotype associated with increased plasma membrane permeability and cell lysis.7 The molecular events responsible for initiating death, however, were not investigated in these studies; consequently, whether this form of cell death shares some features of apoptosis (e.g., mitochondrial depolarization, caspase activation) remains to be determined.Mechanism of the Cell Death Caused by HIV PR
Bcl-2
In 1996, Strack and colleagues first identified Bcl-2 as an apoptosis regulatory protein that is cleaved by HIV PR and suggested that this event results in the infected cell’s death.8,9 Bcl-2 is an important cellular survival protein that inhibits programmed cell death via homo- or heterodimerization with other Bcl-2 family members. This antiapoptotic molecule contains four functional BCR-homology GTPase activation domains (BH-domains) and one membrane target domain (transmembrane [TM] domain) (see Figure 10.1).10,11 All four BH domains are required for the antiapoptotic effects of Bcl-2 to form homodimers and heterodimers.
Specifically, the BH1 to BH4 domains mediate interactions with other Bcl-2 members in which the BH1 to BH3 domains form a hydrophobic groove, and the N-terminal BH4 domain stabilizes this structure.11 The HIV PR cleavage site within Bcl-2 is located between the BH3 and BH1 domains (between phenylalanine 112 and alanine 113).8 This cleavage separates Bcl-2 into two parts: an N-terminal part containing the BH4 and BH3 domains and the C-terminal part containing the BH1, BH2, and TM domains (Figure 10.1). Although the C-terminal part still contains Bcl-2 functional motifs -helix 5 and 6 (pore formation), this fragment is unlikely to confer cell death protection, as it lacks the BH3 domain (ligand domain) and BH4 domain (cell-death protecting domain). The BH3 and BH4 domains are important for the ability of the full length of Bcl-2 to protect cells from death. Specifically, the BH3 domain is necessary for Bcl-2 to bind BH3-containing proapoptotic proteins,12 whereas the BH4 domain is required for interaction with Raf kinases.13 During caspase-3- dependent apoptosis, active caspase-3 removes the BH4 domain of Bcl-2, converting it to a Bax-like proapoptotic effector.14 Therefore, removal of the BH3 and BH4 domains of Bcl-2 by HIV PR cleavage is likely to abrogate the antiapoptotic function of Bcl-2 and may even promote proapoptotic signaling.Procaspase-8
Caspase-8 is an initiator caspase that is required for death receptor-initiated death signaling.15,16 Procaspase-8 is activated after its interaction with the adaptor proteins FADD (Fas-associated protein with death domain) or TRADD (tumor necrosis factor receptor-associated protein with death domain), resulting in the removal of the procaspase N-terminal prodomain by caspase-8 autoprocessing. The activation of this initiator caspase is a key step in physiological apoptosis, and
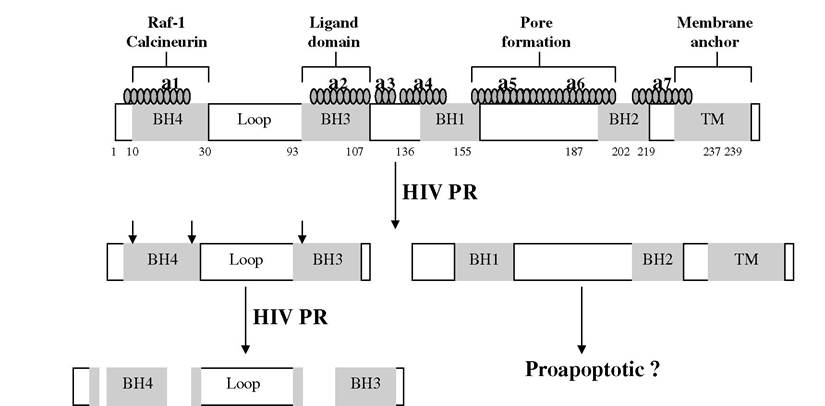
FIGURE 10.1 Human immunodeficiency virus (HIV) protease inhibitor (PR) cleaves Bcl-2.
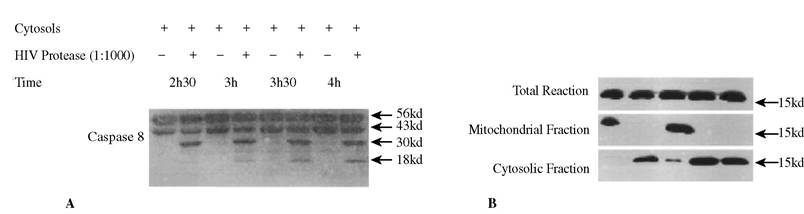
FIGURE 10.2 HIV-1 PR processes and activates procaspase 8 (A) and causes cytochrome c release from mitochondria (B) in cell-free system.
the activation of procaspase-8 may also be modified during virus infections. Some lytic viruses (e.g., Sendai virus) induce apoptosis through activation of caspase-8,17 but several latent viruses, such as -herpesviruses (including Kaposi’s sarcoma-associated human herpesvirus-8) directly inhibit the maturation of procaspase-8 through the death-effector domain (DED) containing viral protein vFlip.18 Recent data suggest that HIV PR may also process procaspase-8. HIV PR processes recombinant procaspase-8 in vitro and cellular procaspase-8 in a cell free system.19 The processing of procaspase-8 by HIV-1 PR leads to cytochrome c release from mitochondria into the cytoplasmic compartment in a comparable manner to the release of cytochrome c seen with recombinant active caspase-8 or Granzyme B (Figure 10.2A, B), resulting in the activation of the downstream caspase cascade. Co-incubation of HeLa nuclei with the activated cytoplasm extracts from Jurkat cells causes HeLa nuclei fragmentation and internucleosomal DNA cleavage (as determined by DNA ladder analysis), which are the classical markers of cell death through apoptosis (Figure 10.3A-E).
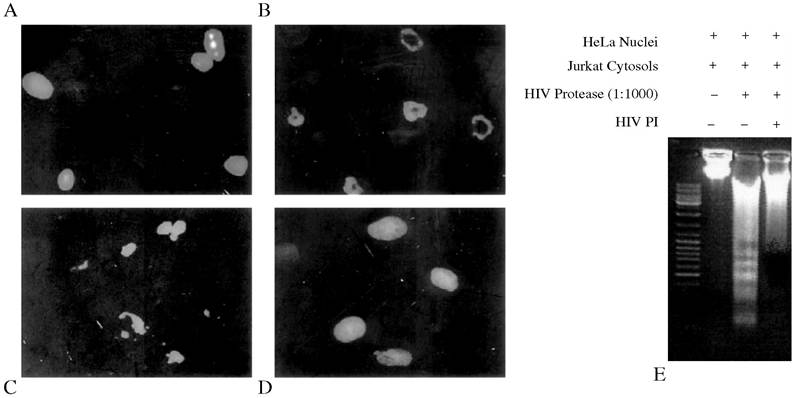
FIGURE 10.3 HIV-1 protease induces the nuclear changes of apoptosis. Jurkat cytosols (1 mg) were treated with recombinant HIV-1 protease at 30°C for 3 hours and then co-incubated with HeLa nuclei. Treated nuclei were imaged under microscopy by Hoechst 33342 staining. (A) Nuclei incubated with Jurkat cytosols without HIV-1 protease treatment. (B) Nuclei incubated with Jurkat cytosols treated with HIV-1 protease, resulting in fragmentation of the nuclear membrane and chromatin condensation or (C) margination of chromatin. (D) The induction of apoptotic changes were completely inhibited by HIV-1 protease inhibitor. (E) HIV-1 protease induces internucleosomal DNA fragmentation. DNA gel from nuclei incubated with Jurkat cytosols in the presence or absence of HIV-1 protease with or without HIV-1 protease inhibitor (PI).
Taken together, HIV-1 PR could activate an apoptotic-signaling pathway via direct targeting of procaspase-8 to induce apoptosis in HIV-infected cells.