HIV-1 TAT AND APOPTOSIS
Tat is produced early after infection and is essential for viral gene expression and virus production. Work conducted in the last few years by several laboratories has shed light on the numerous interconnected functions of this striking small protein, both intra- and extracellularly.
Its extracellular functions play an important role in viral infection. Significant levels of extracellular Tat are found in the serum from HIV-1-infected patients and in Kaposi’s sarcoma lesions, in which it is associated with endothelial spindle cells. It is also released in the culture supernatants of cells infected with HIV-1.30,31 It has been shown with Tat-transfected COS-1 cells that Tat is released in the absence of cell death or changes in cell permeability via a leaderless exocytosic pathway.32,33 During acute infection of T cells by HIV, extracellular Tat released in the stromal microenvironment of infected cells can bind or can be efficiently taken up by most cell types.30’32-35 In infected cells, Tat will then transactivate virus gene expression and replication, and in uninfected cells it will favor the transmission of both macrophage-tropic and T lymphocyte-tropic HIV-1 strains by inducing the expression of the chemokine receptors (and HIV-1 co-receptors) CCR5 and CXCR4.36-38 Specific motifs are engaged in Tat internalization, namely, the basic domain, which interacts with negatively charged sulfated groups of free heparin and heparan sulfates and to cell-surface heparan sulfate proteoglycans, and the RGD motif. Thus, Tat protein, which is readily released from infected cells and taken up by neighboring (infected or uninfected) cells, is implicated in HIV-1 pathogenesis not only by its indispensability for virus replication but also by its numerous other control functions that ultimately affect cell survival, growth, and angiogenesis.By the early 1990s, Tat was shown to inhibit proliferation and induce apoptosis in activated uninfected cells.31,39-41 Depending on the cell type and conditions (in particular, Tat concentrations and source), apoptosis of uninfected “bystander” cells was also described. Tat-dependent apoptosis was described in several cell types, including T and B cells, erythroid cells, endothelial cells, and neuronal cells. For example, Tat was found to display apoptotic properties on CD4+ Jurkat T lym- phoblastoid cells but was ineffective when unstimulated PBMCs or PBMCs activated by anti-CD3 antibodies (OKT3) were incubated with up to 5 μM synthetic Tat Lai or Tat fragments (Figure 9.2). In the case of Jurkat cells, apoptosis was demonstrated by different techniques, including flow
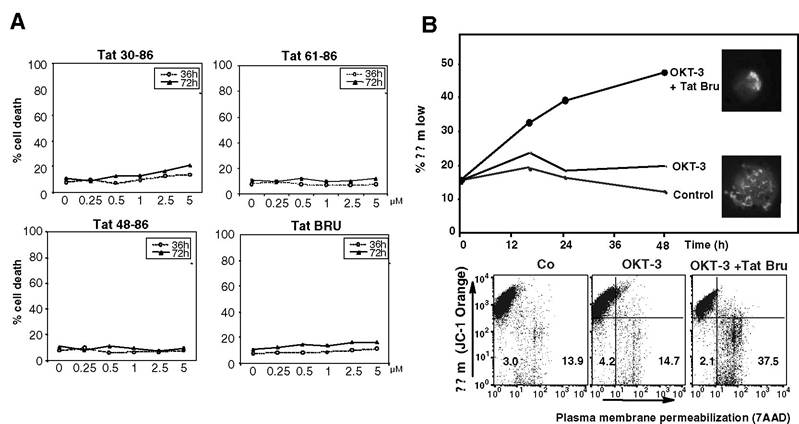
FIGURE 9.2 Tat promotes apoptosis of CD3-stimulated Jurkat cells but does not kill PBMCs. (A) PBMCs were exposed up to 72 h to indicated doses of synthetic Tat protein or Tat fragments, labeled with 7 aminoactinomycin D (7-AAD), and submitted to FACS analysis. Curves are the means of three independent experiments (SD < 5%). Note that, in these experimental conditions, there is no significant cellular uptake of 7-AAD, thus indicating the absence of Tat-induced PBMC death. (B) Jurkat cells were stimulated for 24 h in the presence of OKT3 (anti-CD3), were then submitted or not to synthetic Tat Lai, were colabeled with the ∆Ψm-sensitive probe JC-1 and with the DNA-interacting dye Hoechst 33342 and with 7-AAD, and were submitted to both FACS analysis and fluorescence microscopy observations. A representative ∆Ψm time response (after OKT3 plus Tat treatment) is shown in the upper panel. Inserted fluorescent micrographs show Jurkat cells incubated (upper insert) or not (lower insert) with synthetic Tat (5 μM) for 24 h and stained with 1 μM ∆Ψm-sensitive dye JC-1 (light dots in bottom image fluorescence shows mitochondria with high ∆Ψm, light area on the upper right side of the top image fluorescence shows mitochondria with low ∆Ψm) and with Hoechst 33342 (base color of top and bottom images fluorescence).
Dot plot was obtained by FACS analysis after costaining with JC-1 and 7-AAD.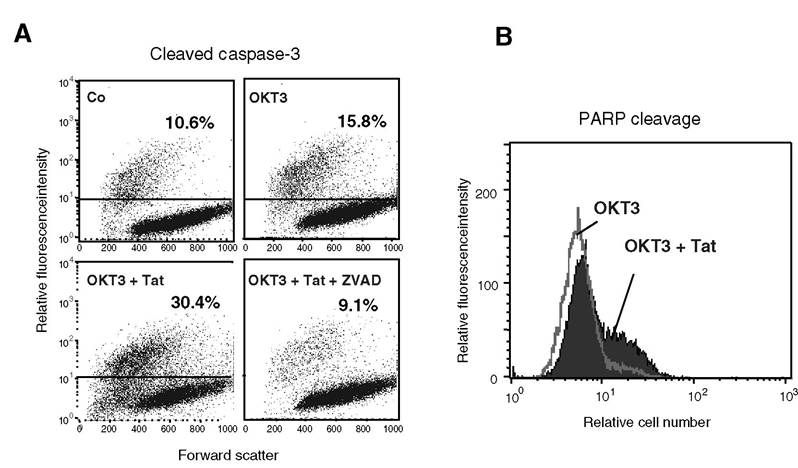
FIGURE 9.3 Tat enhances caspase-3 activation and PARP cleavage in CD3-stimulated Jurkat cells. Experimental conditions were similar to those described in the legend of Figure 9.2. At 24 h post-Tat treatment, cells were subjected to permeabilization and labeled with either a polyclonal FITC-tagged antibody directed to activated caspase-3 (A) or a polyclonal FITC-tagged antibody directed to cleaved PARP (B). A representative experiment is shown. Note that pretreatment with the pan-caspase inhibitor zVAD-fmk (50 μM) fully inhibited Tat-induced caspase-3 activation.
cytometry after propidium iodide labeling of treated cells, the TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick-end labeling) method, the cleavage of caspase-3 and poly(ADP ribose polymer)ase (PARP) revealed by FITC-labeled specific antibodies and FACS analysis, and the evaluation of mitochondrial transmembrane potential (∆Ψm) state (Figure 9.2 and Figure 9.3)42 (Lecoeur and Jacotot, personal communication). Tat was also shown to induce cell death by apoptosis in primary cortical neurons (Lecoeur, Chauvier et al.43) and cultured human fetal neurons44 and rat neurons. For example, it was shown recently with striatal neurons that both HIV-1 gp120 and Tat fragment 1-72 significantly increased caspase-3 activation in a concentrationdependent manner, but that even though both components induced significant neurotoxicity, the nature of the apoptotic pathways preceding death differed.45
Mechanisms by which Tat was reported to induce apoptosis include upregulation of the apop- totic effector molecule Fas ligand in a pathway that might involve NF-κB,31,46 inhibition of the expression of manganese-dependent superoxide dismutase (Mn-SOD),47 and the activation of cyclin-dependent kinases.41 These studies were performed in the context of uninfected cells.
Thus, in both T cells and HeLa cells, it was shown, for example, that Tat suppressed the expression of Mn-SOD, a mitochondrial enzyme that is part of the cellular defense system against oxidative stress.47 The truncated Tat fragment 1-72, known to transactivate the HIV-1 long terminal repeat (LTR), was unable to affect Mn-SOD expression, the cellular redox state, or TNF-mediated cytotoxicity. (TNF is known to stimulate HIV-1 replication through activation of NF-κB.) This result highlights the importance of the C-terminal end of Tat protein in this process.Tat binds tubulin and polymerized microtubules through a four-amino-acid residue subdomain located in the core region, leading to the alteration of microtubule dynamics and activation of a mitochondria-dependent apoptotic pathway.48
Tat-dependent apoptosis may also be explained by the effects of Tat on Bcl-2 and related proteins. Tat-mediated downregulation of Bcl-2 was effectively observed at both the transcriptional and translational levels. Also, Tat-transfected cells expressed increased amounts of Bax, a Bcl-2 family protein known to induce apoptosis.49 In contrast, however, stable and transient transfections of Jurkat cells with the cDNA of Tat and a plasmid containing Bcl-2 promoter in front of CAT (Bcl-2 Pr/CAT) were found to stimulate CAT activity and lead to an increase of Bcl-2 mRNA and protein expression. This effect was specifically related to Tat, because Jurkat cells transfected with the cDNA of Tat in antisense orientation, Tat carrying a mutation at the position 22 (mutant C22G), or the control vector alone (pRPneo-SL3) did not show any significant difference in Bcl-2 promoter activity with respect to parental Jurkat cells.50
In the context of infected cells, Tat was found to induce apoptosis by upregulating caspase-8, a protease that cleaves downstream caspases such as caspase-3, thus initiating the caspase cascade and increasing the sensitivity of cells to apoptotic signals.51 The induction of apoptosis could be prevented by treating cells with the caspase-8 inhibitor 2-IETD-fmk.
The capacity of Tat to upregulate caspase-8 was shown in both CD4+ T cells and T cell lines, and it was found that Tat does not require Fas-Fas ligand interaction to induce apoptosis. It was demonstrated further that Tat transactivation mutants, namely, Tat C22G and Tat K41A mutants, induced apoptosis, whereas a truncated Tat fragment 1-72 did not. This result supports the fact that the C-terminal of the protein (expressed by the second exon) is required to induce apoptosis in the context of a viral infection and that induction of apoptosis by Tat is a function independent from transactivation.51 One report proposed that Tat may accumulate at the mitochondria in stressed Tat-expressing cells, and this mitochondrial localization was found to be correlated with the ∆Ψm disruption detected in these cells.52Controversies remain, however, because Tat can either induce apoptosis31,39-49,51-53 or inhibit apoptosis, depending on context.50,54,55 Furthermore, certain cells, such as chimpanzee T cells, have been shown to be resistant to Tat-enhanced apoptosis. A number of studies were devoted to the molecular mechanisms by which Tat might directly or indirectly regulate apoptosis. Several cofactors mediating Tat-induced apoptosis were characterized, and models of Tat-mediated apoptosis were proposed. At this stage, however, it seems apparent that the observed ability of Tat to activate or inhibit apoptosis is highly dependent on the cell type, its stage of activation and infection, the use of tumoral or primary cells, their environments (presence of co-stimulatory signals and other cells), and the method used to reveal apoptosis.54,56,57 Significant differences were also observed in the presence of recombinant and synthetic Tat according to their integrity, their degree of purity, and their proper folding. The length (86 versus 106 residues) of the protein might also be important. This suggests that the results of some studies would need to be revisited.