INTRODUCTION
Multicellular organisms rely on a fine balance between cell proliferation and cell death for their health. Alterations in this balance inevitably lead to loss of tissue homeostasis and the onset of several diseases.
More than 30 years ago, Kerr and coworkers described a novel form of cell death with peculiar morphological characteristics, and its crucial role in cellular biology became clear in the following years.1 They named this type of cell death “apoptosis.” Apoptosis is a highly organized and genetically controlled type of cell death, which has proven to be essential during embryonic development to ensure proper organogenesis.2 It is characterized by a number of sequential morphological alterations, such as chromatin condensation, cell shrinkage, plasma membrane alterations with surface blebbing, and, in the latest stages, fragmentation of the cell into membrane-bound vesicles containing intact organelles called apoptotic bodies.3 Removal of the apoptotic bodies is achieved via phagocytosis by neighboring cells and professional phagocytes such as macrophages. The prompt clearance of dying cells before the onset of secondary necrosis and lysis of the apoptotic bodies supposedly prevents the triggering of the immune response.Apoptosis is also crucial in adult organisms, as it enables the elimination of unwanted aged, harmful, or infected cells to maintain normal cellular homeostasis and preserve tissue functions. Insufficient apoptosis can result in development of cancer or autoimmunity, whereas excessive cell death has been linked to acute injuries (such as stroke; myocardial infarct; fulminant hepatitis; reperfusion damage), chronic degenerative diseases (including multiple sclerosis, amyotropic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease), immunodeficiency (including acquired immunodeficiency syndrome [AIDS]); and infertility.
the Apoptotic machinery
Although several different stimuli can trigger apoptosis, the apoptotic signaling within the cell occurs mainly via two defined molecular pathways: the death receptor pathway (also called the extrinsic pathway) and the mitochondrial pathway (also called the intrinsic pathway). The extrinsic pathway is initiated by the engagement of a family of cell surface cytokine receptors named death receptors, and it generates an apoptotic signal that may or may not require the involvement of mitochondria for its execution. The intrinsic pathway is triggered by different external or internal signals that culminate in mitochondrial dysfunction, such as irradiations, oxidative stress, or some chemotherapeutic drugs. The mitochondrial dysfunction is characterized by disruption of mitochondrial architecture and function and release of proapoptogenic factors from the organelle (Figure 2.1). Both the intrinsic and the extrinsic pathways rely on a group of proteolytic enzymes called caspases (cysteinyl aspartate-specific proteases) as central executors of the cell death program. Indeed, activated caspases are responsible for the degradation of several cellular substrates that results in the morphological and biochemical alterations characteristic of the apoptotic phenotype. Endogenous inhibitors of caspases (i.e., inhibitors of apoptosis proteins [IAPs]) are present in the cell to prevent accidental caspase activation. Other proteolytic enzymes, such as calpain and the lysosomal cathepsins, can be activated during apoptosis, but their role in the apoptotic program has not been completely defined and seems to be dependent on cell type and nature of the apoptotic stimulus.4
Mitochondrial dysfunction in apoptosis is regulated by the Bcl-2 family of proteins, which includes both pro- and antiapoptotic members. These proteins are perhaps the most important regulators of the mitochondrial pathway, serving upstream and at the level of the mitochondria to integrate pro- and antiapoptotic signals.
The balance between pro- and antiapoptotic members of the family, as well as their reciprocal interactions, determines whether the mitochondrial pathway of apoptosis is initiated or not. Although the extrinsic and the intrinsic pathways were initially thought to be independent, the BH3-only protein Bid has been found to provide a cross talk between the two pathways. Indeed, Bid is activated after death receptor engagement and translocates to the mitochondria, where it triggers the mitochondrial pathway of apoptosis. The above-mentioned components of the apoptotic machinery and their main endogenous inhibitors are here discussed in greater detail.Caspases
The signaling process by either the extrinsic or intrinsic pathway involves the activation of caspases. Caspases are cysteine proteases that cleave their substrates on the carboxyl side of aspartic acid residues. Their role as central executioner of cell death has been amply documented in the last decade5 and is supported by the evidence that caspases seem to be evolutionary conserved, being identified in Drosophila melanogaster, Caenorhabditis elegans, and Xenopus laevis, as well as in mammals. To date, 14 mammalian caspases have been identified, 12 of which have also been cloned in humans (Table 2.1). Caspases are constitutively and ubiquitously expressed as inactive proenzymes (zymogens) that require proteolytic processing for acquiring catalytic activity. The proenzyme possesses a variable-length N-terminal prodomain, a large subunit containing the active site cysteine in a conserved QACXG pentapeptide motif, and a C-terminal small subunit. By sequential processing of two Asp cleavage sites, the prodomain is removed, and the large and small subunits associate to provide the active form of the enzyme, a tetramer composed of two heterodimers.6 Their specificity to cleave substrates on the carboxy-terminal side of an Asp residue, together with their need to be proteolytically activated by cleavage at Asp sites, renders the caspases capable of self-activation as well as of activating each other in a cascade-like process.
The mammalian caspases can be divided into two families: those primarily involved in apoptosis (caspase-2, -3 -6, -7, -8, -9, -10, and -12) and those mainly involved in inflammatory processes and cytokine processing (caspase-1, -4, -5, and -11). Based on the length of their prodomains, the caspases involved in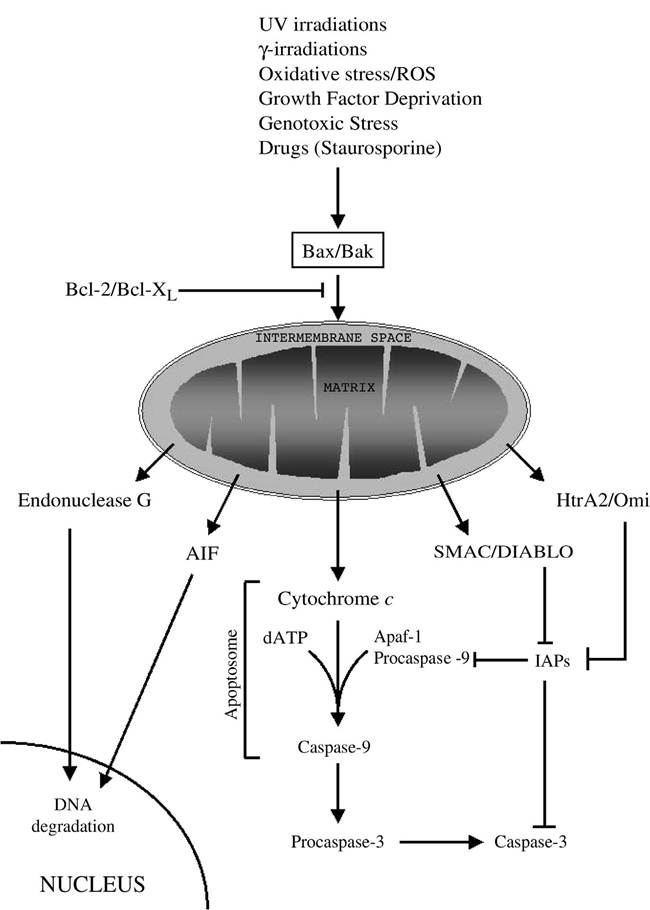
FIGURE 2.1 The mitochondrial/intrinsic pathway of apoptosis. Different stimuli, including UV and γ-irradi- ation, growth factor deprivation, oxidative stress with production of reactive oxygen species (ROS), genotoxic stress, and several drugs, trigger the intrinsic pathway via the activation of proapoptotic members of the Bcl-2 family of protein (i.e., Bax, Bak), which oligomerize on the outer mitochondrial membrane and cause mitochondrial dysfunction. The proapoptotic action of Bax and Bak can be antagonized by the antiapoptotic members of the same family Bcl-2 or Bcl-XL. After mitochondrial dysfunction, several apoptogenic factors, including cytochrome c, Smac/DIABLO, Htr2A/Omi, AIF, and endonuclease G, are released from the mitochondrial intermembrane space. Cytochrome c binds to Apaf-1 and procaspase-9 to form a complex named apoptosome, which, in an ATP-requiring reaction, results in the activation of the initiator caspase-9. Caspase-9, in turn, activates the effector caspases (caspase-3, -6, and -7) responsible for the degradation of cellular substrates. Smac/DIABLO and Htr2A/Omi contribute to caspase activation by binding and inactivating the endogenous inhibitor of caspase IAPs. AIF and endonuclease G translocate to the nucleus, where they induce DNA degradation.
TABLE 2.1
Classification of Mammalian Caspases
| Name | Human Homolog | Prodomain | Adaptor | Protein-Protein Interaction Motifs | Function |
| Caspase-1 | Yes | Long | CARDIAK | CARD | Inflammation |
| Caspase-2 | Yes | Long | RAIDD/CRADD | CARD | Apoptosis (initiator) |
| Caspase-3 | Yes | Short | Apoptosis (effector) | ||
| Caspase-4 | Yes | Long | Unknown | CARD | Inflammation |
| Caspase-5 | Yes | Long | Unknown | CARD | Inflammation |
| Caspase-6 | Yes | Short | Apoptosis (effector) | ||
| Caspase-7 | Yes | Short | Apoptosis (effector) | ||
| Caspase-8 | Yes | Long | FADD | DED | Apoptosis (initiator) |
| Caspase-9 | Yes | Long | Apaf-1 | CARD | Apoptosis (initiator) |
| Caspase-10 | Yes | Long | FADD | DED | Apoptosis (initiator) |
| Caspase-11 | Yes | Long | Unknown | CARD | Inflammation |
| Caspase-12 | Yes | Long | Unknown | CARD | Apoptosis |
| Caspase-13 | No | Long | Unknown | CARD | Apoptosis |
| Caspase-14 | No | Short | Apoptosis (effector) |
apoptosis can be further divided into either upstream caspases (also known as initiator or apical caspases) or downstream caspases (also called effector or distal caspases).
Upstream caspases (caspase-2, -8, -9, and -10) generally have long prodomains containing caspase-recruitment domain (CARD) or death-effector domain (DED) motifs that mediate the recruitment to caspase-activating adaptors (such as Fas-associated protein with death domain [FADD] or apoptotic activating factor1 [Apaf-1]). The long prodomain also promotes self-association of procaspase molecules, resulting in their autocatalytic activation. On the contrary, downstream caspases (caspase-3, -6, and -7) lack the ability to self-associate and do not possess CARD motifs due to the short prodomains; therefore, they require cleavage by initiator caspases for their activation. Activated downstream caspases are responsible for the degradation of several cellular substrates associated with the morphological changes of apoptosis, including DNA degradation, chromatin condensation, and membrane blebbing.
Bcl-2 Family
The Bcl-2 family of intracellular proteins arbitrates the life-or-death decision in response to diverse physiologic and cytotoxic stimuli. The balance between its anti- and proapoptotic members regulates the mitochondrial integrity and determines the susceptibility of the cell to mitochondria-mediated apoptosis. For this reason, the Bcl-2 family is considered a critical, if not the most important, regulator of the mitochondrial pathway of apoptosis (also referred to as the intrinsic pathway).
Bcl-2 (B-cell lymphoma-2), the first member discovered and the prototype of the family, was cloned from the t(14;18) break point in human follicular lymphoma as a proto-oncogene with antiap- optotic properties.7 Since then, a number of proteins structurally similar to Bcl-2 have been discovered, and they are generally referred to as the Bcl-2 family. To date, the mammalian Bcl-2 family comprises at least 20 members sharing various degrees of homology within four conserved regions named Bcl-
2 homology (BH) 1-4 domains.8 The family can be further divided into three main subclasses, defined, in part, by this homology and, in part, by their functions (Table 2.2).
A first subclass includes proteins with the highest homology to Bcl-2 in all the four conserved BH 1-4 domains. Members of this group are Bcl-2, Bcl-XL, Mcl-1, A1, and Bcl-w, all of which strongly inhibit apoptosis in response to many cytotoxic insults. They are generally localized on the mitochondrial outer membrane, but some haveTABLE 2.2
The Bcl-2 Family
| Class | Name | BH Domains | TM | Function in Apoptosis | Organism |
| Bcl-2 like | Bcl-2 | BH 1-4 | Yes | Inhibits | Mammalian |
| BcI-Xl | BH 1-4 | Yes | Inhibits | Mammalian | |
| Bcl-Xs | BH 3-4 | Yes | Promotes | Mammalian | |
| Bcl-W | BH 1-4 | Yes | Inhibits | Mammalian | |
| Mcl-1 | BH 1-4 | Yes | Inhibits | Mammalian | |
| A1∕Bfl-1 | BH 1-4 | No | Inhibits | Mammalian | |
| Boo/Diva | BH 4,1-2 | Yes | Inhibits | Mammalian | |
| CED-9 | BH 4,1-2 | Yes | Inhibits | C. elegans | |
| E1B 19K | BH 1 | No | Inhibits | Adenovirus | |
| BHRF-1 | BH 1-2 | Yes | Inhibits | Epstein-Barr virus | |
| KS-Bcl-2 | BH 1-2 | Yes | Inhibits | Human herpesvirus-8 | |
| ORF-16 | BH 1-2 | Yes | Inhibits | Herpesvirus Saimiri | |
| LMW5-HL | BH 1-2 | No | Inhibits | African swine fever virus | |
| Bax-like | Bax | BH 1-3 | Yes | Promotes | Mammalian |
| Bak | BH 1-3 | Yes | Promotes | Mammalian | |
| Bok | BH 1-3 | Yes | Promotes | Mammalian | |
| BH3-only | Bad | BH 3 | No | Promotes | Mammalian |
| Bid | BH 3 | No | Promotes | Mammalian | |
| Bik | BH 3 | Yes | Promotes | Mammalian | |
| Blk | BH 3 | Yes | Promotes | Mammalian | |
| Hrk | BH 3 | Yes | Promotes | Mammalian | |
| BNIP3∕Nix | BH 3 | Yes | Promotes | Mammalian | |
| Bim/Bod | BH 3 | Yes | Promotes | Mammalian | |
| Bmf | BH 3 | ? | Promotes | Mammalian | |
| Puma | BH 3 | ? | Promotes | Mammalian | |
| Noxa | BH 3 | No | Promotes | Mammalian | |
| EGL-1 | BH 3 | No | Promotes | C. elegans |
been found also on the endoplasmic reticulum and the nuclear membrane. The mechanisms by which they prevent mitochondrial dysfunction are still largely unclear. Nonetheless, Bcl-2 has been found to inhibit activation and oligomerization of the proapoptotic member Bax.9 A second subclass, the so- called Bax-like proteins, includes the multidomain proapoptotic members of the family that share homology in the BH 1-3 domains. Members of this group are Bax, Bak, and Bok. Whereas Bok expression seems to be limited to reproductive tissues, Bax and Bak are widely distributed. In healthy cells, Bax is found in the cytosol as a monomer, but after an apoptotic stimulus, it undergoes conformation changes, integrates into the outer mitochondrial membrane, and oligomerizes.10-12 Bak is an oligomeric integral mitochondrial membrane protein, but it also undergoes conformation changes during apoptosis and forms larger aggregates.12,13 The third subclass comprises the BH3-only members, proapoptotic proteins containing only the BH3 domain. Members of this group are Bid, Bik, Bad, Hrk, Bim, Bmf, Noxa, and Puma. They act as sensors to promote apoptosis in response to proximal death signals or intracellular damage, generally (but not exclusively) by binding to and neutralizing their prosurvival relatives. To prevent unnecessary triggering of apoptosis, in healthy cells, they are subjected to tight regulation either by transcriptional control or posttranslational modification or cellular compartmentalization. Bid requires proteolytic cleavage to be fully activated after death receptor engagement. Bad activation and inactivation in response to growth and survival factors are regulated by its phosphorylation status and by sequestration in the cytosol through binding to 14-3-3 proteins. Noxa and Puma are transcriptionally regulated by p53 in response to DNA damage. Bim and Bmf are sequestered on the cytoskeleton by binding to dynein light-chain complexes 1 and 2 in the microtubular dynein motor complex (Bim) and actin filamentous myosin V motor complex (Bmf), and they can be activated by multiple stimuli.
Both types of proapoptotic proteins are required to initiate apoptosis: specific death signals result in the activation of BH3-only proteins, which, in turn, activate the multidomain proapoptotic members Bax or Bak either by directly interacting with them14 or by binding to the antiapoptotic Bcl-2-like proteins and antagonizing their inhibitory functions.15 Activation of either Bax or Bak triggers mitochondrial dysfunction and is required for apoptosis.16 Indeed, inactivation of either Bax or Bak does not result in any severe phenotypic alterations, but simultaneous inactivation of both genes dramatically impairs apoptosis in many tissues.16,17
Inhibitors of Apoptosis
Because all cells contain the apoptotic machinery required for cell death, it is clear that cells must also have inherent inhibitory mechanisms to control for inappropriate activation of the machinery and to ensure survival. Over the past several years, cellular mechanisms and proteins have been identified that function at various points in the cell death process to prevent or inhibit uncontrolled activation of the death program. A critical checkpoint of apoptosis exists at the level of the mitochondria, which is mainly regulated by the Bcl-2 family of proteins, as described earlier in this chapter. Here, we report on endogenous inhibition of apoptosis at another crucial point, the activation of caspases.
Inhibitors of Apoptosis Proteins (IAPs)
The inhibitors of apoptosis proteins (IAPs) constitute a family of proteins that function as intrinsic regulators of the caspase cascade. To date, IAPs are the only endogenous proteins identified that regulate the activity of both initiator (caspase-9) and effector caspases (caspase-3 and -7). IAPs are characterized by one or more baculoviral IAP repeat (BIR) domains, cysteine- and histidine- rich protein folding domains of approximately 70 amino acids that chelate zinc.18 However, because not all BIR-containing proteins have been associated with cell death, IAPs have been defined as BIR-containing proteins (BIRPs) with antiapoptotic properties.19 The other members of the BIRPs family generally contain only a single BIR domain and function mainly in cytokinesis and chromatin segregation. Members of the mammalian family of IAPs include, among others, neuronal apoptosis inhibitory protein (NAIP), cellular IAP-1 and IAP-2 (c-IAP1 and c-IAP2), X-linked IAP (XIAP), and survivin. The members c-IAP-1, c-IAP-2, and XIAP contain three BIR domains and a carboxy terminal RING finger20,21; c-IAP-1 and c-IAP-2 also contain a CARD domain near their C-termini, but this seems to not be essential for the functional activity of the proteins.22 Several IAPs, including XIAP, c-IAP-1, and c-IAP-2, bind directly to caspase-3, -7, and -9, but not to other caspases, such as caspase-1, -6, and -8.22 In particular, it seems that IAPs with multiple BIR motifs bind to procaspase-9 through the third BIR domain and to the active forms of caspase-3 and -7 through the second BIR domain.22 By targeting procaspase-9 and inhibiting its activation, the IAPs block the mitochondrial pathway of apoptosis, whereas by targeting active caspase-3 and -7, they block those stimuli that can bypass the mitochondria and directly target the effector caspases. The RING-finger domain of XIAP and c-IAP-1 can trigger the ubiquitination and degradation of both caspase-3 and -723 and IAP proteins in response to apoptotic stimuli.24 This apparent contradiction can be explained by different cellular responses to various degrees of apoptotic stress. The RINGfinger may function to suppress apoptosis in conditions of low apoptotic stimulus (via ubiquitination and proteasome-mediated degradation of caspases), whereas higher levels of apoptotic stress may trigger self-degradation of the IAPs and cell death.
Whereas the inhibitory effect of IAPs is crucial in healthy cells to prevent aberrant activation of caspases, this effect must be relieved when the cell is signaled to undergo apoptosis. This process is mediated by a mitochondrial protein named second mitochondria-derived activator of caspases (Smac)25 or direct IAP-binding protein with low pI (DIABLO).26 Smac usually resides in the mitochondrial intermembrane space and is released into the cytosol after an apoptotic stimulus, together with cytochrome c. Whereas cytochrome c contributes to the apoptotic cascade by directly activating Apaf-1 and caspase-9, Smac interacts with multiple IAPs and relieves their inhibitory effect on both initiator and effector caspases. Smac binds to both the BIR2 and BIR3 domains of XIAP and, possibly, of c-IAP-1 and c-IAP-2. The conserved IAP-binding motif in Smac competes with that in caspase-9 for the binding to IAPs and thus releases caspase-9 from inhibition by IAPs. A motif located between BIR1 and BIR2 on XIAP is responsible for binding and inhibiting caspase- 3 or -7.
FLICE/Caspase-8-Inhibitory Proteins (FLIPs)
Death receptor-mediated caspase activation can be modulated by a family of proteins called the FLIPs. FLIPs were first identified in viruses (v-FLIPs) as proteins with antiapoptotic activity containing two tandem DEDs that enable them to bind to the Fas death-inducing signaling complex (DISC) and to other death receptor DISCs, and to block caspase-8 activation.27 A human cellular homologue was later identified and called c-FLIP (also known as FLAME-1, I-FLICE, or Casper, or CASH, or MRIT, or CLARP, or Usurpin).28 c-FLIP exists in short- and long-splicing variants. The short form, c-FLIPS, consists only of two DEDs and is similar in structure to v-FLIP. The long form, c-FLIPl (also called I-FLICE), resembles caspase-8 and caspase-10 in its structure, and it consists of two DEDs along with an inactive caspase-like catalytic domain, where the active site cysteine residue embedded in the conserved pentapeptide QACRG or QACQG motif present in all caspases is replaced by a tyrosine. For this reason, c-FLIPL shows no cysteine protease activity. Both forms of c-FLIP are thought to be recruited to the DISC on stimulation through interaction with their death domain (DD).29 FLIPs compete with the caspases for binding to the adaptor FADD. Whereas c-FLIPs may directly compete with procaspase-8 for binding to the DISC, c-FLIPl allows the recruitment and initial cleavage of procaspase-8 to the DISC but prevents its complete proteolytic processing to generate the active subunits.30 Displacement or inhibition of caspases by FLIPs prevents the onset of the proteolytic caspase cascade and blocks death-receptor-mediated apoptosis.