MOLECULAR PATHWAYS OF APOPTOSIS
Several stimuli that trigger apoptosis seem to do so by inducing activation of proteolytic enzymes— in particular, but not exclusively, caspases. In order to activate initiator caspases, several molecules of caspase zymogens have to be brought to close proximity to allow self-processing thanks to the moderate catalytic activity of the zymogens.
This is achieved through specific adaptor proteins that facilitate the recruitment of initiator caspases into large complexes. Activated upstream caspases then initiate a proteolytic cascade culminating in activation of effector caspases, degradation of cellular substrates, and cell death. Here, we describe the main signaling pathways that promote initiator caspase activation and apoptosis following a variety of stimuli.The Mitochondrial Pathway (The Intrinsic Pathway)
A large variety of different stimuli, including ultraviolet or gamma irradiation, oxidative stress, growth factor deprivation, and several cytotoxic compounds including kinase inhibitors such as staurosporine, have been reported to trigger caspase activation and apoptosis via a mitochondrial pathway (Figure 2.1).31 Although very different in nature, all these stimuli result in activation of proapoptotic members of the Bcl-2 family, which tightly regulates the mitochondrial function. The multidomain proapoptotic proteins, such as Bax and Bak, can be activated directly, or indirectly, via interaction with BH3-only proteins of the same family. For example, Bid is cleaved by caspase-8 to generate a C-terminal fragment (t-Bid) that interacts with Bax and Bak, triggering activation of these proteins and facilitating their insertion into the mitochondrial membrane, possibly through destabilization of the membrane. When activated, Bax and Bak permeabilize the outer mitochondrial membrane, causing the release of proapoptotic proteins, such as cytochrome c, Smac/DIABLO, HtrA2ZOmi, apoptosis-inducing factor (AIF), and endonuclease G, from the intermembrane space.
The mechanism(s) by which Bax or Bak induce mitochondrial permeabilization are still debated. Studies on artificial liposomes suggested that Bax and Bak oligomers might form channels on the outer mitochondrial membrane.32 Alternatively, it has been proposed that Bax might interact with components of the permeability transition (PT) pore, such as the voltage-dependent anion channel (VDAC), on the outer mitochondrial membrane to create larger channels or to induce pore opening with subsequent mitochondrial swelling and membrane rupture.9,33,34 After they enter the cytosol, the proapoptogenic proteins released from the mitochondria promote either caspase activation or DNA degradation via multiple signaling pathways (Figure 2.1). Cytochrome c binds to the adaptor Apaf-1, an adenosine triphosphate (ATP)-binding protein. Complex formation with cytochrome c increases Apaf-1 affinity for deoxyadenosine triphosphate (dATP) and induces conformational changes in the protein leading to oligomerization and exposure of a CARD domain. Through the CARD domain, Apaf-1 recruits procaspase-9 to form a complex referred to as the apoptosome.35 The proximity of several molecules of procaspase-9 results in its proteolytical autoactivation and in the subsequent activation of downstream effector caspases, such as caspase-3 and -7, via a cascade mechanism. The simultaneous release of SmacZDIABLO and HtrA2ZOmi potentiates the mitochon- dria-mediated activation of caspases. SmacZDIABLO binds to the IAPs in the cytosol and antagonizes their inhibitory effect on both initiator caspases (i.e., caspase-9) and effector caspases (i.e., caspase-3 and -7). HtrA2ZOmi, a serine protease, contains an N-terminal amino acid sequence almost identical to the N-terminal sequence of SmacZDIABLO and binds and inactivates IAPs in a way similar to SmacZDIABLO.36 HrtA2ZOmi serine protease activity also seems to be associated with its ability to activate caspases, but the mechanism is still largely unknown.37 A second group of mitochondrial proapoptogenic factors contributes to the execution of the apoptotic program by promoting DNA degradation. AIF translocates from the cytosol to the nucleus after being released from the mitochondria.38,39 Studies performed in cell-free systems demonstrated that, after entry into the nucleus, AIF induces chromatin condensation and large-scale DNA fragmentation, with no requirement for additional cytosolic factors.38 Recombinant AIF, in the presence of cytosol, has also been reported to act on mitochondria with a feedback mechanism to induce disruption of the membrane potential and release cytochrome c. Both of these effects are caspase independent. Similar to AIF, endonuclease G also translocates to the nucleus after being released from the mitochondria, where it induces DNA fragmentation in a caspase-independent manner.40,41The Death Receptor Pathway (The Extrinsic Pathway)
The death receptors are a subset of the tumor necrosisZnerve growth factor (TNFZNGF) receptor superfamily. Members of the death receptor family include, among others, Fas (also called CD95 or APO-1), tumor necrosis factor-receptor 1 (TNF-R1, also called p55 or CD120a), and tumor necrosis factor-related apoptosis-inducing ligand-receptors 1 and 2 (TRAIL-R1 and TRAIL-R2, also called DR4 and DR5). When activated through interaction with their natural ligands, a group of complementary cytokines that belongs to the TNF family of proteins, these receptors are able to trigger an apoptotic signaling cascade.42-44 The members of this family are all type-I transmembrane proteins with a cysteine-rich, extracellular N-terminal domain, a membrane-spanning region, and a C-terminal intracellular tail containing a sequence of approximately 80 amino acids (the DD). The DD allows for the recruitment of adaptor molecules, which function as “docking proteins” to bind the proform of the initiator caspase-8, -10, and, perhaps, -2.
Signal transduction by death receptors is initiated by the oligomerization of the receptor that follows the engagement of the ligand to the receptor’s extracellular domain (Figure 2.2).
As a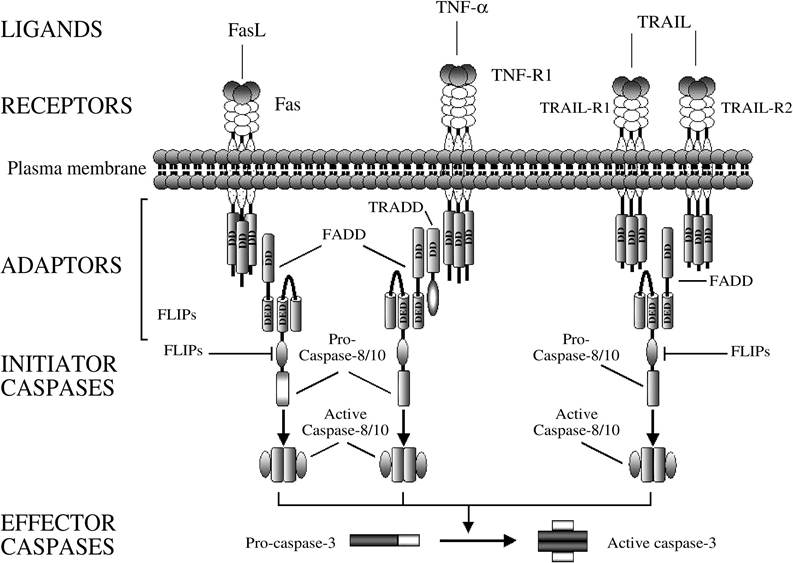
FIGURE 2.2 The death receptor/extrinsic pathway of apoptosis. Examples of signaling through the main death receptors (Fas/CD95/APO-1, TNF-R1, TRAIL-R1, and TRAIL-R2) are illustrated here. Engagement of death receptors by their cognate ligands results in oligomerization of the receptor and recruitment of adaptor proteins. The death domain (DD) on the adaptor protein interacts with the receptor’s DD, whereas the death effector domain (DED) binds the correspondent DED in the prodomain of the inactive initiator caspase-8 or -10. The resulting complex is referred to as the death-inducing signaling complex (DISC). The proximity of several procaspase molecules results in the activation of the caspase by self-processing, which, in turn, activates downstream caspases such as caspase-3. The inhibitors of apoptosis cFLIPs prevent the recruitment or inhibit the processing of procaspase-8 to the DISC.
consequence, different DD-containing adaptor proteins, such as FADD or tumor necrosis factor receptor-associated protein with DD (TRADD), are recruited to the receptor through homophilic interaction of the receptor’s and adaptor’s DDs. Some adaptor proteins also contain a DED that mediates the recruitment of caspases through the association with correspondent DED or CARD motifs in the prodomain of the inactive initiator caspases. The resulting complex is referred to as the DISC. The proximity of several procaspase molecules recruited to the receptor results in self-processing and activation of the caspase, which subsequently starts a cascade of caspase activation by directly processing and activating the effector caspases (i.e., caspase-3, -6, and -7). With regard to Fas, this type of signaling is referred to as “type I” and is generally associated with high levels of caspase-8 recruited and activated at the DISC (Figure 2.3).45 Alternatively, the activation of effector caspases may occur indirectly, via a mitochondria-mediated process, with a signal referred to as “type II.” In type-II cells, the amount of caspase-8 activated at the DISC seems to be insufficient to directly cleave effector caspases, but it can efficiently cleave the BH3-only protein Bid to generate a fragment that translocates to the mitochondria and induces mitochondrial dysfunction.45-47 After permeabilization of the outer mitochondrial membrane, several proapoptotic factors are released in the cytosol, where they contribute to caspase activation and apoptosis, as previously described for the mitochondrial pathway.
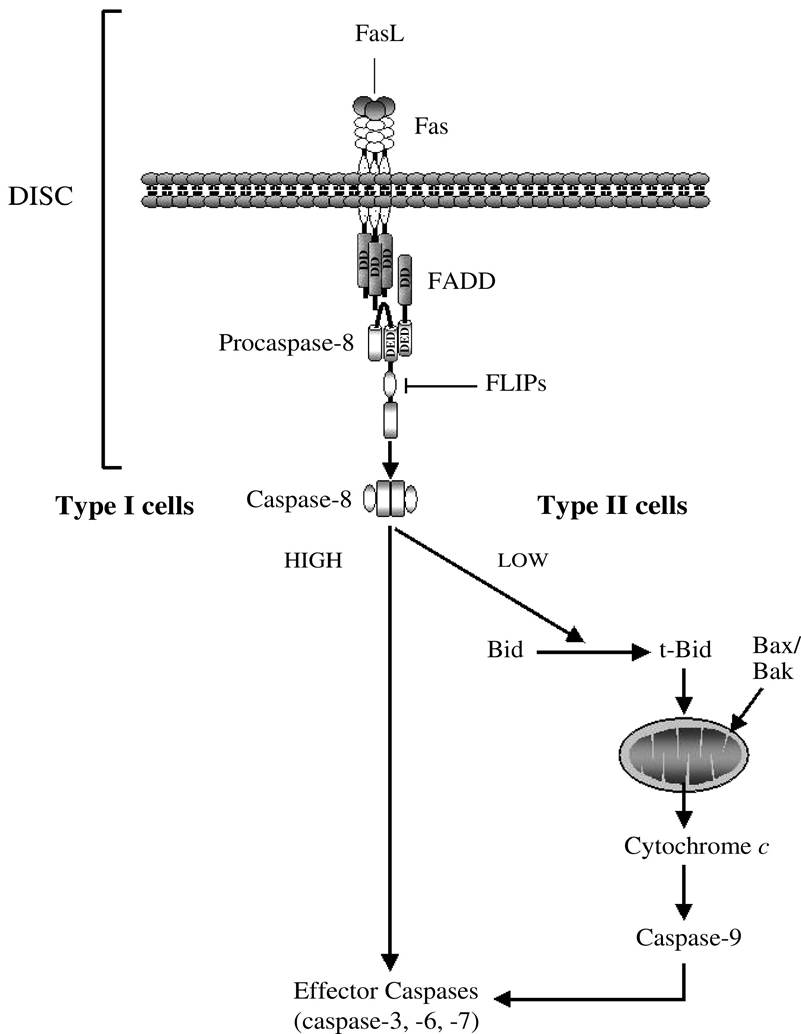
FIGURE 2.3 Fas∕CD95-mediated apoptotic pathways in type I and type II cells. The amount of active caspase- 8 generated at the DISC determines whether the cell activates a mitochondrial-independent (type I) or mitochondrial-dependent (type II) pathway of caspase activation and apoptosis.