A VIEW TO A KILL—GENERAL MECHANISMS OF APOPTOSIS
Programmed cell death is required for immune system function, tumor surveillance, and prevention of autoimmune diseases. Apoptosis was originally defined by morphological features, including cell shrinkage, membrane blebbing, nuclear condensation, and chromatin margination.
Death is the consequence of an orchestrated series of biochemical events leading to the controlled destruction of the dying cell. Biochemical hallmarks, notably DNA fragmentation as seen by “laddering,” loss of asymmetric distribution of phosphatidylserine (PS) in the plasma membrane, disruption of mitochondrial function, and degradation of key cellular proteins, indicate that death has occurred by an apoptotic mechanism.initiator caspases
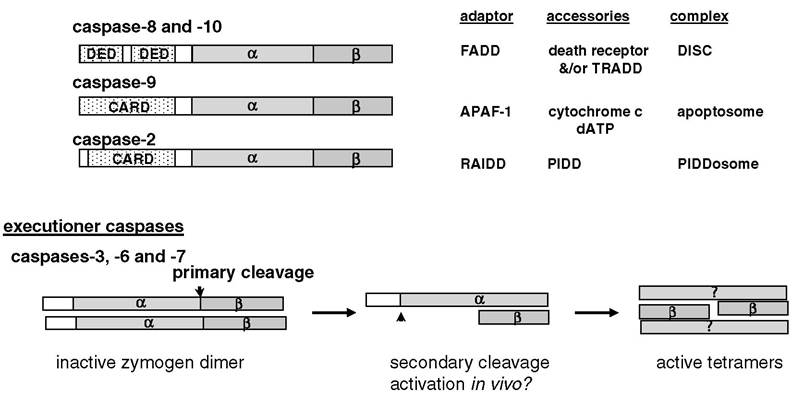
FIGURE 5.1 Overview of mammalian apoptotic caspases. The initiators are characterized by a long prodomain that contains interaction motifs (death effector domain [DED] or caspase-recruitment domain [CARD]) responsible for intermolecular homotypic interactions with adaptor molecules, leading to enzymatic activation. Cleavage between the large (α; ~p20) and small (β; ~p10) subunits does not appear to be necessary for activation, and removal of the prodomain may serve to stabilize the active protease in vivo. Activation of the executioner caspases occurs following interchain cleavage between the large (α) and small (β) subunits. Subsequent autocatalytic removal of the short prodomain may be required for protease activation in vivo.
The pressures upon a cell to die depend on developmental stage, activation state, and environment. There is a wide variety of cell-specific and context-dependent death stimuli, and there are a number of common features shared between apoptotic mechanisms at play in the immune system.
Morphological features that define apoptosis are mediated by members of the caspase family of intracellular proteases. Caspases are a family of cysteine proteinases that cleave substrates at the C-terminal side of aspartate residues (i.e., Asp at P1 of the cleavage site). Additional specificity is imparted by residues at P4, P3, and P1 of the substrate cleavage site for each respective member of the caspase family.1,2 The best characterized caspases involved in apoptosis can be divided into two subgroups based on genetic and functional attributes: initiator and executioner caspases (see Figure 5.1). All known caspases are expressed as single polypeptide chains that are present in the cell as inactive zymogens. The initiators are the first caspases to be activated during the induction of phase of apoptosis. Initiator caspases (caspase-8, -10, -9, and -2) are characterized by long prodomains that possess CARD (caspase-recruitment domain) or DED (death-effector domain) motifs responsible for homotypic interactions with similar motifs in their corresponding adaptor partners. Initiator caspases are activated by homodimerization facilitated by an adaptor protein that acts as a scaffold upon which the initiator caspase becomes activated. Cleavage between the large and small subunits of the initiators may serve to stabilize the proteolytically active enzyme complex in vivo.The executioner caspases (caspase-3, -7, and -6) are responsible for bringing about the morphological features that define apoptosis. The executioners are also expressed as inactive zymogens; however, they are activated by cleavage of the zymogen polypeptide between the large (α) and small (β) subunits. With one exception, the only known proteases known to cleave and activate executioner caspases are other caspases. The exception is the serine protease granzyme B expressed primarily by cytotoxic lymphocytes. Cleavage between the large and small subunits is followed by autoprocessing to remove the prodomain, which is substantially shorter than the prodomains found in initiator caspases and is devoid of CARD or DED motifs.
The conformation of the resulting active executioner caspase is (αβ)2 (see Figure 5.1).In 1982, Wylie, an early pioneer of apoptosis research, used glucocorticoid treatment of thymocytes to demonstrate what has become recognized as a hallmark of apoptosis: DNA laddering as seen by nucleosomal-sized fragments by agarose gel electrophoresis in the final stages of cellular destruction.3 The deoxyribonuclease (DNase) responsible for causing the signature DNA laddering was not discovered until 1998, when Nagata and colleagues identified the caspase-activated DNase (CAD).4’5 CAD is coexpressed with its inhibitor ICAD. Cleavage of inhibitor of CAD (ICAD) by caspase-3 overcomes the inhibitory activity of ICAD, which, in turn, releases the active CAD nuclease. The coexpression of a proapoptotic protein along with its own inhibitor is a recurring theme in apoptosis. A likely explanation is that it is dangerous for a cell to express a death-inducing protein without additional regulatory constraints. Any leakiness in such a system would be rapidly selected out.
DNA fragmentation represents the end stage of cell death, as there is little chance for recovery after a cell has lost its genome. In most cases, the nucleus is not essential to initiate apoptosis (except by genotoxic stress), but because most cells contain nuclei, it is clear that degradation of DNA is necessary at some point during programmed death. It was speculated that the nucleases that degrade cellular DNA could also be co-opted to degrade DNA associated with an infecting virus in cells undergoing apoptosis induced by cytotoxic lymphocytes. Although this is an appealing notion, it is not yet supported by direct experimental evidence.
Another hallmark of apoptosis is the loss of asymmetry in the plasma membrane. Phosphatidylserine (PS) is restricted to the inner leaflet of the plasma membrane in healthy cells but becomes evenly distributed during apoptosis.
Loss of membrane asymmetry, known as PS-flip, has provided a convenient indicator of apoptosis for assays in vitro.6 More importantly, PS-flip contributes to the recognition of apoptotic cells by phagocytes in vivo, which, in turn, is involved in the immunosuppressive response observed upon macrophage engulfment of apoptotic cells. Extracellular PS exposure is one of the engulfment signals sent by apoptotic cells.7Immune regulation and homeostasis rely heavily on the ability to induce death by apoptosis. Removal of the dead cell avoids the deleterious effects of a disseminated inflammatory response. Early models suggested that inflammation was avoided simply by preventing spillage of cellular contents. Recent developments, however, suggest a more complex mechanism, whereby dying cells secrete cytokines and induce the secretion of immunosuppressive cytokines from the phagocytic cells. One of the mediators of this anti-inflammatory effect has been identified as the PS-receptor (PSR). PSR is one of a growing number of recognition receptors that bind molecular flags presented on the external surface of apoptotic cells. Engagement of these receptors facilitates the uptake and removal of dying and dead cells, and the receptors also signal the production of cytokines.8-10 These findings are consistent with earlier observations that secretion of TGF-β and IL-10 by macrophages in response to apoptotic cells contributes to the maintenance of an extracellular immunosuppressive milieu. Interestingly, dying cells can contribute to suppression of inflammation. In addition to having the decency of not dumping their contents, apoptotic cells were shown to induce expression of IL-10 and release it from existing intracellular stores.
Apoptotic and necrotic cells elicit different immunostimulatory effects on antigen-presenting cells (APCs). Engulfment of apoptotic cells resulted in better presentation of cellular antigen in the context of both major histocompatibility complex (MHC) classes I and II than did engulfment of necrotic cells, but necrotic cells elicited much stronger APC activation.11,12 The result of this response may be that apoptotic cells are presented efficiently but elicit only a limited response due to the low level of danger, whereas a greater immune response results from the presence of necrotic cells due to the heightened possibility of danger.