12 Congenital Anomalies
Jill Berkin
Melissa L. Russo
Congenital anomalies are among the most common causes of neonatal morbidity and mortality. According to the Centers for Disease Control and Prevention, they occur in 3% of live births and account for 25% of all pediatric hospital admissions.
Birth defects can involve an isolated organ system or multiple organ systems; multiple anomalies may encompass a syndrome. The causes of congenital anomalies are genetic, environmental, or unknown etiology. There are multiple risk factors that have been associated with increased risk of congenital anomalies.ETIOLOGY
• Causes of congenital anomalies may be chromosomal, familial, multifactorial, or idiopathic; hence, obtaining a thorough family history and screening of low-risk populations are important.
• Genetic etiologies include the following: chromosomal disorders such as trisomy (e.g., Down syndrome), deletion (e.g., DiGeorge syndrome), or monosomy (e.g., Turner syndrome); monogenic disorders such as Noonan syndrome and Smith-Lemli-Opitz syndrome; and multifactorial disorders such as isolated congenital heart disease, cleft lip and palate, and arthrogryposis which result from interactions of several genes and environmental factors.
• NongeneticZenvironmental etiologies include the following teratogens: ethanol, certain medications such as tretinoin (Retin-A) and warfarin (Coumadin), illicit
P.165 drugs, maternal nutritional deficiencies, and maternal medical conditions such as diabetes and maternal infections such as toxoplasmosis or syphilis (see Chapter 11).
TABLE 12-1 Factors Associated with Increased Risk for Congenital Abnormalities
Advanced maternal age (maternal age ≥35 years at time of delivery)
Pregestational diabetes
Exposure to a known teratogen
History of having a child with birth defect
Personal or family history of a known genetic abnormality (e.g., balanced translocation, mutation, or aneuploidy)
Abnormal serum screening
Multiple gestation
Assisted reproductive technology
• Ninety percent of infants with congenital anomalies are born to women with no risk factors (T able 12-1).
SCREENING AND MANAGEMENT
• Given the significant morbidity and mortality of congenital defects, all patients should be offered screening for fetal chromosomal abnormalities preferably during the first trimester and a level II anatomy ultrasound at 18 to 22 weeks. Detailed ultrasonography by an experienced technician can detect up to 80% of fetal anomalies, allowing the full range of management options: expectant management, in utero therapy, further workup (e.g., karyotyping, microarray and/or viral studies), and pregnancy termination.
• Management should include counseling that takes into consideration the fetus, the mother, and the family. Treatment options and prognosis should be discussed. With a fetal congenital anomaly, multidisciplinary approach facilitates a unified plan of care. The obstetrician or maternal fetal medicine (MFM) specialist can coordinate care with genetic counselors, neonatologists, and other pediatric specialists such as surgeons, cardiologists, urologists, and neurosurgeons. Social work and bereavement counseling can also be part of the care plan if indicated. The care plan must be timely, unbiased, and sensitive to the concerns and values of the patient and her family.
• Ultrasonography can be used to diagnose many major anomalies. The other clinical uses for ultrasound entail confirmation of gestational age, definition of placental location, determination of amniotic fluid volume, and evaluation of fetal growth.
• Optimal timing for the anatomic survey is between 18 and 20 weeks' gestation. At this gestational age, organogenesis is complete, bony ossification in the skull does not yet obscure sonography, and structures are large enough for accurate assessment but still small enough to visualize within a single ultrasound window. With a detected anomaly, a patient can pursue a genetic workup and has a full set of options available to her at the time the anomaly is discovered.
P.166
• The structures that are assessed in the level II anatomy screen include the following:
• Head: The biparietal diameter and head circumference are measured, both in the same view at the level of the thalamus and cavum septum pellucidum.
The intracranial contents, ventricular structures, cerebellar diameter, and cisterna magna are evaluated.• Spine: Sagittal, transverse, or coronal views are obtained at all levels to screen for neural tube defects.
• Heart: Four-chamber view and visualization of left and right outflow tracts are required. If an abnormality is suspected, fetal echocardiography should be performed.
• Abdomen: The stomach and umbilical vein should be visualized in the same plane for the abdominal circumference measurement. Abdominal wall defects are ruled out by verifying normal cord insertion and the absence of bowel loops in the amniotic fluid. The kidneys, renal pelvises, and bladder are evaluated for location, structure, and evidence of obstruction.
• Limbs: The four limbs should be imaged to their distal ends and the humerus and femur measured. The hands should be seen to open and close and the feet examined for normal positioning and appearance.
• Several sonographic “soft markers” occur more frequently in fetuses with aneuploidy, specifically trisomy 21. These markers include increased nuchal translucency, renal pelvis dilation, echogenic intracardiac focus
(small bright spot within the fetal heart on ultrasound), echogenic bowel, and short long bones. Aneuploidy risk increases with an increased number of markers identified; previous studies have reported likelihood ratios for the individual markers.
CHROMOSOMAL ABNORMALITIES WITH ASSOCIATED CONGENITAL ANOMALIES
In many specific chromosomal syndromes, there are characteristic findings prenatally that assist in prenatal diagnosis. Common aneuploidies will be discussed with their associated findings (Table 12-2A, 12-2B).
T risomy 21 (Down syndrome)
• Down syndrome is the most common aneuploidy where the fetus survives, with a frequency of 1:660 to 1:800 births. With trisomy 21, there is an extra copy of chromosome 21. The frequency of nondisjunction increases with increasing maternal age.
• Down syndrome can be complete trisomy 21 in which all cells have three copies of chromosome 21 (94% of cases) or mosaic trisomy 21 in which only some cells in the body have an abnormal number of chromosome 21 (2% to 3%).
A third etiology of Down syndrome results from a mother who has a balanced translocation, in which an extra piece of chromosome 21 is attached to another chromosome and is given to the fetus.• The finding of an echogenic intracardiac focus is not an indication for a fetal echocardiogram, as this is not a structural defect, but should prompt a search for other markers of Down syndrome and a discussion of a potential increased risk in the pregnancy. If Down syndrome is suspected, a fetal echocardiogram is recommended, as these fetuses have a higher incidence of congenital heart defects.
• Children with Down syndrome have some degree of intellectual disability, and in prenatal counseling, it is important to discuss that there is a spectrum of disease and the severity of disease cannot be predicted prenatally or by genetic testing.
P.167
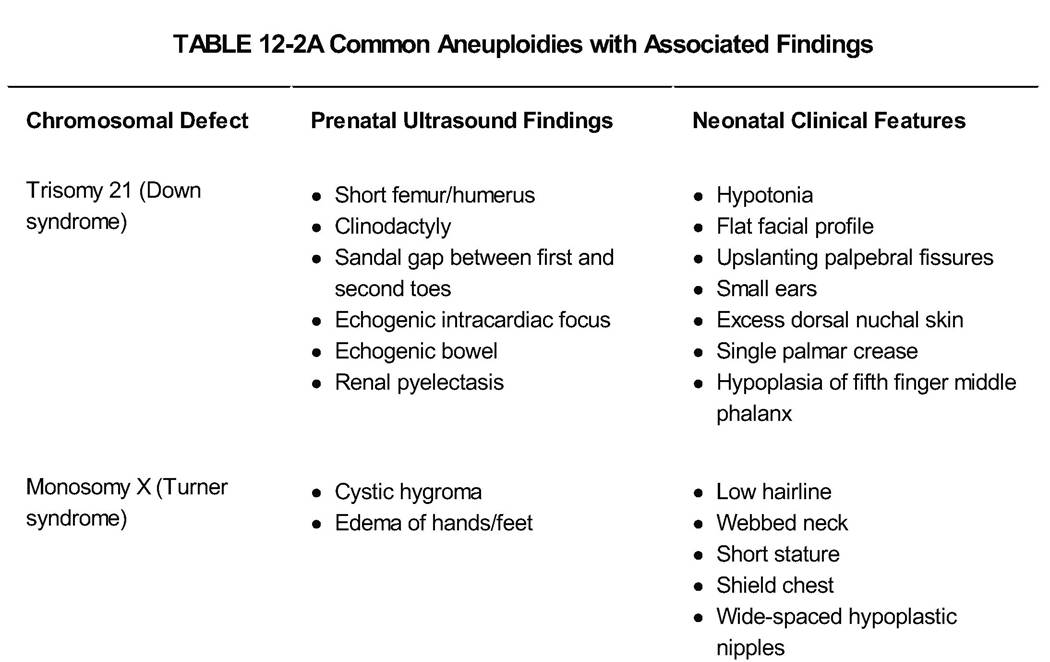

TABLE 12-2B Common Aneuploidies with Associated Findings
| Chromosomal Defect | Neonatal Findings |
| Trisomy 13 (Patau syndrome) | Holoprosencephaly; cardiac defects; hypotelorism; abnormalities of orbits, nose, and palate; abnormal ears, omphalocele, polycystic kidneys, radial bone aplasia, skin aplasia, polydactyly |
| Trisomy 18 (Edwards syndrome) | IUGR; cardiac defects; prominent occiput; rotated and malformed ears; short palpebral fissures; small mouth; clenched hands with second and fifth fingers overlapping third and fourth fingers; horseshoe kidney; radial bone aplasia; hemivertebrae; imperforate anus |
IUGR, intrauterine growth restriction.
P.168
Trisomy 13 and 18
• Trisomy 13 (Patau syndrome) is usually due to meiotic primary nondisjunction giving rise to a 47 +13, XX or XY genotype. T risomy 13 is invariably fatal; approximately 50% of newborns die in the first month of life and 90% die by 1 year.
Of those that survive, they have multiple anomalies and severe intellectual disability.• Trisomy 18 (Edwards syndrome) is most commonly due to meiotic primary nondisjunction giving rise to a 47
+18, XX or XY genotype. Life expectancy for these infants is usually very limited.
T urner Syndrome
• Turner syndrome (monosomy X) is usually 45, X genotype. Some individuals are mosaic, with both 45, X and 46, XX cell lines, with resultant variable characteristics. These individuals can have some degree of learning disability.
Triploidy
Triploidy has one extra haploid set of chromosomes (i.e., 69 chromosomes). Most cases are 69, XXY (60%) or 69, XXX (37%). Only 3% of cases are 69, XYY. T riploidy is uniformly fatal within the first few months of life.
COMMON SPECIFIC CONGENITAL ANOMALIES
Congenital Heart Abnormalities
Congenital heart abnormalities are among the most common birth defects (Table 12-3). There are some known etiologies for congenital heart disease (CHD), namely maternal diabetes, teratogen exposure, and certain genetic causes such as 22q11 microdeletion (i.e., DiGeorge syndrome). There is a long-established association between congenital heart defects and aneuploidy. The frequency of chromosomal abnormalities with a congenital heart defect has been estimated to be 16% to 45% prenatally and 5% to 10% postnatally; the discrepancy in these rates is secondary to antenatal death occurring in fetuses with chromosomal abnormalities. The likelihood of a cardiac defect exceeds 50% for Down syndrome and is 90% for trisomies 13 and 18.
• Prenatal diagnosis of CHD has increased secondary to advances in ultrasound resolution and fetal echocardiography. A fetal echocardiogram is recommended for any abnormality detected in the standard heart views and for any fetus at high risk of a congenital heart defect (e.g., diabetic mother, exposure to a teratogen in the first trimester, previous child with CHD). Some CHDs need a higher level of surveillance during the pregnancy to watch for signs of fetal heart failure in utero.
Hydrops in utero is a poor prognostic sign.• The functional consequences of cardiac anomalies are usually not evident until conversion from fetal to neonatal circulation after birth. Some common defects, such as ventriculoseptal defects and coarctation of the aorta, can be missed on prenatal ultrasounds and fetal echocardiograms.
• Management depends on the specific type of cardiac defect. Prenatal management entails offering genetic counseling secondary to the association of a chromosomal or genetic etiology for the CHD, the option of prenatal diagnosis with amniocentesis, appropriate pediatric cardiology, and pediatric cardiac surgery consultation. Most cardiac defects can be corrected surgically, although multiple procedures are usually required. Secondary to the complex nature of these cases, delivery at a tertiary care center is recommended.
P.169
TABLE 12-3 Most Common Congenital Cardiac Defects
| Name | Percentage of Cardiac Defectsa | Findings | ||
| Hypoplastic left heart syndrome (HLHS) | 2%-4% | Small left ventricle, aortic atresia, hypoplastic mitral valve | ||
| Endocardial cushion defect/atrioventricular septal defect (AVSD) | 2%-7% | Missing “crux” of the heart in four-chamber view | ||
| Ventricular septal defect (VSD) | 20%-40% | Abnormal communication between left and right ventricles, causing shunt | ||
| Persistent truncus arteriosus | 1%-2% | Single overriding arterial trunk | ||
| T ransposition of great arteries (TGA) | 2.5%-5% | Aorta arises from right ventricle and pulmonary artery from left. | ||
| Double outlet right ventricle (DORV) | 1%-2% | Both great arteries arise from right ventricle. | ||
| Tetralogy of Fallot (TOF) | 3%-7% | VSD, overriding aorta, pulmonary artery stenosis, right ventricular hypertrophy | ||
aDoes not add to 100%; minor cardiac defects are not listed. Adapted from Woodward PJ, Kennedy A, Roya S, et al, eds. Diagnostic Imaging: Obstetrics, 1st ed. Salt Lake City, UT: Amirsys/Elsevier, 2005.
Neural T ube Defects
Neural tube defects (NTDs) are congenital structural abnormalities of the brain and spine and are the second most common form of structural congenital anomalies. NTDs result from failure of neuropore closure during the third and fourth weeks of gestation. They occur in 1 to 2 per 1,000 births. The main forms of NTD are anencephaly and spina bifida (Table 12-4). Spina bifida can be closed or open and there are different types. NTD risk factors include family history of NTD, poorly controlled diabetes, seizure medications, and poor nutritional status or low folate stores.
• Prevention with preconception folate supplementation (0.4 mg/day) significantly lowers the incidence of NTDs. For women with a previously affected pregnancy, a higher dose of 4.0 mg of daily folate is recommended.
• Prenatal screening with maternal serum alpha-fetoprotein (MSAFP) should be offered to all pregnant women in the second trimester. The alpha-fetoprotein (AFP) is elevated in 89% to 100% of pregnancies complicated by NTDs and an abnormal value is defined as more than 2.5 times the normal.
• Prenatal diagnosis can be made by ultrasound with confirmation by amniocentesis for AFP and acetylcholinesterase levels. The prenatal ultrasound shows splaying of dorsal vertebral elements and a meningeal sac. Other intracranial findings are the “lemon sign” from scalloping of the frontal bones and the “banana sign” of the compressed cerebellum. Ventriculomegaly is also common along with Arnold-Chiari II abnormalities.
P.170
TABLE 12-4 Types of Neural Tube Defects
| Anencephaly | Spina Bifida | |
| Ultrasound findings | î Absent cranial vault î Absent telencephalon and encephalon î Polyhydramnios | î Vertebral splaying +/- overlying soft tissue î Lemon sign î Banana sign |

• Management of NTDs entails delivery at a tertiary care center where neonatology and neurosurgery are available. Delivery at term is preferred. The mode of delivery is determined on an individual basis; however, there have been no improved outcomes with C-section for these fetuses. In terms of when to repair this defect, the Management of Myelomeningocele Study trial recently compared prenatal versus postnatal closure and found that the children who had prenatal surgery had some improved outcomes but with an increased risk of preterm delivery and uterine dehiscence at delivery.
Hydrocephalus and Ventriculomegaly
Hydrocephalus is a pathologic increase in intracranial cerebrospinal fluid (CSF) volume; hydrocephalus results from fluid production that exceeds absorption or primary atrophy of brain parenchyma. The majority of cases are secondary to an obstruction at some level.
Ventriculomegaly is a descriptive term of enlarged ventricles and this finding can be secondary to various causes. Some of these include obstruction, inability to resorb CSF, maldevelopment of a ventricle or agenesis of a structure in the brain, ex vacuo destructive phenomenon from cerebral atrophy, fetal aneuploidy, genetic such as X-linked hydrocephalus, or infections such as cytomegalovirus or toxoplasmosis. A less common cause of ventriculomegaly is cerebral hemorrhage. If this diagnosis is being considered, a workup for neonatal alloimmune thrombocytopenia (NAIT) should be offered (see Chapter 20).
• Prenatal diagnosis of enlarged ventricles on ultrasound is the definitive finding. The fetal biparietal diameter may or may not be increased with the finding of enlarged ventricles. The method for assessing ventricular size is the atrial diameter measured in a transverse view. If the mean diameter is greater than 10 mm, this indicates the presence of ventriculomegaly. The incidence of associated anomalies with enlarged ventricles varies from 54% to 84%; thus, it is important to have targeted sonographic exam to ensure there are no other abnormalities. Management of the pregnancy with enlarged ventricles includes a determination of the cause. The diagnostic studies include amniocentesis for karyotype, DNA analysis for L1CAM mutations, and
P.171 viral studies. A multidisciplinary team that includes perinatologists, genetic counselors, pediatric neurosurgery, and neonatology should be involved in the care of this pregnancy. Pregnancy termination may be considered in some cases. Fetuses with ventriculomegaly should be delivered at a tertiary care center where a pediatric neurosurgery team is available. The mode of delivery is individualized, and if there is no significant head enlargement, a vaginal delivery is reasonable. Significant head enlargement may preclude vaginal delivery and may be an indication for early delivery.
• Prognosis for ventriculomegaly depends on etiology, severity of ventriculomegaly, and the presence of associated abnormalities. The degree of ventricular dilation does not appear to be associated with poor longterm outcome.
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia (CDH) is a failure of the diaphragm to fuse properly during embryologic development, resulting in abdominal contents occupying the thoracic cavity. This creates a mass effect that prevents normal lung development and leads to pulmonary hypoplasia and persistent pulmonary hypertension, with significant morbidity and mortality in the newborn. Approximately 1 in 2,000 to 3,000 newborns have CDH. Hernias are more often unilateral, posterior, and left sided.
• Prenatal diagnosis of CDH is accomplished in 60% to 90% of cases by ultrasonography or fetal magnetic resonance imaging. On ultrasound, abdominal contents (stomach, bowel, and/or liver) are seen in the thoracic cavity. Other signs seen on ultrasound are mediastinal shift, polyhydramnios, and abnormal cardiac axis. Associated structure anomalies are found in approximately 40% of cases, and the most common anomalies are congenital heart defect, renal anomalies, central nervous system anomalies, and gastrointestinal anomalies.
• Management of the pregnancy includes a detailed ultrasound, to ensure there are no other associated abnormalities, and a fetal echocardiogram. Secondary to an association with chromosomal anomalies and some genetic syndromes, amniocentesis should be offered for karyotype and array. The option of termination of pregnancy should be discussed in some cases depending on gestational age and parental choice. A multidisciplinary team that involves neonatology, pediatric surgery, MFM, and genetic counselors can help the patient and her family determine a treatment plan. Delivery should be performed at a tertiary center where pediatric extracorporeal membrane oxygenation (ECMO) is available.
• Prognosis has significantly improved in recent years due to advances in techniques of ventilation and ECMO. Overall survival now exceeds 80%.
Congenital Cystic Adenomatoid Malformation and Bronchopulmonary Sequestration Congenital cystic adenomatoid malformation (CCAM) and bronchopulmonary sequestration (BPS) are two common congenital lung malformations in the differential for a fetal chest mass. CCAM is a cystic malformation of pulmonary tissue and BPS is a mass of nonfunctioning lung tissue that does not communicate with the bronchial tree. These two abnormalities can exist in isolation or as a hybrid of the two lesions. The distinguishing feature is that CCAM typically has a pulmonary blood supply, whereas BPS has blood supply from anomalous systemic vessels.
• Prenatal diagnosis is possible for both lesions. In CCAM, prenatal ultrasound shows a lung tumor that may be cystic or solid and there is an absence of systemic vascular flow. In BPS, an echodense triangular area of tissue is seen, with a systemic feeding vessel on color Doppler sonography. Additional ultrasound findings with BPS are pleural effusion, mediastinal shift, hydrops, and polyhydramnios.
P.172
• Management of pregnancy with either of these lesions entails a detailed ultrasound examination to ensure there are no other anomalies. With both of these lesions, fetal echocardiography should be performed because of an increased incidence in congenital heart defects. In CCAM, the association with chromosomal abnormalities is uncertain; however, amniocentesis is offered to exclude these abnormalities. In BPS, amniocentesis should be offered for fetal karyotype, as chromosomal anomalies have been described with this anomaly. These fetuses should be delivered at tertiary care centers, and prenatal consultation with MFM, pediatric surgery, and neonatology is recommended. These pregnancies need to be serially monitored by an MFM specialist with ultrasound to watch for signs of progression or regression or an associated fetal complication such as hydrops. In the postnatal period, if these lesions persist, surgical excision is usually recommended.
• Prognosis is generally good for fetuses with CCAM and BPS. The long-term outcome of infants with CCAM following resection is excellent. The long-term outcome from BPS depends on the location of the lesion
(intrathoracic vs. extrathoracic) and the degree of pulmonary hypoplasia.
Gastroschisis and Omphalocele
Gastroschisis and omphalocele are the two classic diagnoses for abdominal wall defects that are detected in utero. Gastroschisis is an isolated abdominal wall defect where the herniated abdominal contents have no covering membrane. Omphalocele is a defect in the abdominal wall in which a membrane of peritoneum and amnion covers the herniated abdominal contents. Differences between these two abdominal wall defects are reviewed in Table 12-5.
• Prenatal screening is accomplished by measuring AFP in the second trimester; both of these defects are associated with an elevated MSAFP.
• Prenatal diagnosis is usually made by ultrasound. Gastroschisis is not associated with an increased risk for aneuploidy. Omphalocele has a high incidence of
P.173 associated malformations and chromosomal abnormalities. The option of amniocentesis for fetal karyotype and genetic testing should be offered in the case of omphalocele. Additionally, there is an increased incidence of congenital heart defects in cases of omphalocele, and fetal echocardiography is recommended.
| TABLE 12-5 Omphalocele and Gastroschisis | ||
| Omphalocele | Gastroschisis | |
| Umbilical cord location | Umbilical cord enters into the center of the sac. | Umbilical cord inserts to left of the defect. |
| Physical findings | î Covered by membrane î Varies in size î Contains bowel loops +/- liver | î No covering î Varies in size î Small bowel +/- liver |
| Additional anomalies | Common (up to 45%) | Usually isolated Increased risk for IUGR Intestinal atresia in 10%-15% |
| Associated chromosomal abnormality | bgcolor=white>Up to 30% with chromosomal abnormalityNo association with chromosomal problem | |
| IUGR, intrauterine growth restriction. | ||
• Management in pregnancy entails serial ultrasound assessments to follow the amount and type of abdominal contents that are herniated. A multidisciplinary team should be involved including MFM, genetic counselors, neonatology, and pediatric surgery. Delivery at a tertiary care center enables optimal care of the newborn. The mode of delivery can be vaginal in most cases, if other standard obstetric indications are met.
• Prognosis of infants with abdominal wall defects depends on whether this is an isolated anomaly, other
anomalies, or an underlying chromosomal anomaly.
Renal Anomalies
Congenital renal anomalies can be diagnosed in the prenatal period, including renal agenesis, multicystic dysplastic kidney disease, infantile polycystic kidney disease, and hydronephrosis secondary to ureteropelvic junction obstruction and outlet obstruction.
• Renal agenesis can be unilateral or bilateral. Unilateral renal agenesis has a normal prognosis and there is usually compensatory hypertrophy of the contralateral side. A portion of patients with unilateral renal agenesis have contralateral vesicoureteral reflux. Bilateral renal agenesis is diagnosed after 18 weeks' gestation, as the fetal kidneys do not contribute to the majority of amniotic fluid until after this gestation. On prenatal ultrasound of these fetuses, the kidneys and bladder are not visualized. This condition causes severe oligohydramnios or anhydramnios and is lethal secondary to severe pulmonary hypoplasia.
• Multicystic dysplastic kidney disease (MCKD) is a severe renal abnormality characterized by increased renal size and numerous large noncommunicating cysts alternating with areas of increased echogenicity on ultrasound. Because of the size of the kidney and the numerous cysts, this is usually detected by prenatal ultrasound. MCKD is usually unilateral. In almost half of cases, the contralateral kidney has other malformations, the severity of which determines the overall prognosis. There is also an association with other nongenitourinary anomalies and some genetic syndromes. Amniocentesis should be offered during a prenatal consultation. With unilateral MCKD, prenatal pediatric urology consultation is recommended. Bilateral multicystic dysplasia is associated with severe oligohydramnios and is fatal secondary to pulmonary hypoplasia.
• Polycystic kidney disease encompasses two inherited disorders with diffuse involvement of both kidneys. The so-called infantile polycystic kidney disease (autosomal recessive polycystic kidney disease [ARPKD]) is inherited in an autosomal recessive fashion. From the perspective of prenatal diagnosis and neonatal presentation, the recessive polycystic kidney disease is much more common. This disease is characterized by bilateral, enlarged, echogenic kidneys. Oligohydramnios can be present. Autosomal dominant polycystic kidney disease (ADPKD) rarely presents in the prenatal period; this disease usually has clinical findings in the third or fourth decade of life. Sonogram reveals enlarged kidneys with multiple cysts. Individuals with this form of polycystic kidney disease also have liver cysts, pancreatic cysts, and intracranial aneurysms. Renal ultrasound is recommended in both parents to evaluate for ADPKD. The main cause of perinatal morbidity and mortality is pulmonary hypoplasia. Aggressive neonatal management has lead to 1-year survival rates of 82% to 85% in ARPKD. If infants survive the first month of life, they are predicted to live for many years.
P.174
• Ureteropelvic junction (UPJ) obstruction is the most common cause of significant neonatal hydronephrosis. UPJ obstruction prevents urinary flow from the renal pelvis to the ureter. Most cases are unilateral; bilateral cases have a worse prognosis. Pregnancy management is generally unchanged in unilateral cases, but with bilateral UPJ obstruction, a fetal intervention of urinary shunting may be necessary. There is an overall increased incidence of chromosomal abnormalities with obstructive uropathy; thus, the option of amniocentesis for prenatal karyotype should be offered. A consultation with a pediatric surgeon or pediatric urologist should be offered to the patient as well. With isolated UPJ obstruction, the prognosis is usually favorable.
• Bladder outlet obstructions have the potential to affect the entire urinary and pulmonary system. In males, the most common cause of bladder outlet obstruction is posterior urethral valves (PUVs). In females, the most common cause is urethral atresia. The characteristic prenatal finding on ultrasound is a dilated bladder (megacystis) and bilateral hydroureteronephrosis. With PUVs, the bladder wall is thickened and the urethra may have a characteristic keyhole appearance. There is an association with chromosomal abnormalities; thus, amniocentesis and fetal karyotype should be offered in cases of bladder outlet obstruction. In utero interventions may be helpful in some cases, but there is often severe irreversible renal impairment. Pediatric urology consultation should be offered prenatally. Prognosis is determined
P.175 by evaluation of amniotic fluid volume. The degree of oligohydramnios determines the extent of pulmonary hypoplasia and the ultimate outcome for the fetus.
TABLE 12-6 Skeletal Dysplasias
| Type of Dysplasia and Gene Mutation | Description | Outcome |
| Achondroplasia FGFR3 | î Rhizomelic shortening of limbs î Frontal bossing î “Collar hoop” sign: rounded metaphyseal epiphyseal interface at the femur | Normal intelligence comorbidities: î Joint problems î Craniocervical junction problems î Obstructive sleep apnea î Middle ear dysfunction î Kyphosis |
| Thanatophoric dysplasia FGFR3 | î Very short extremities î Platyspondyly-Aattened vertebral ossification centers î Small chest î Telephone receiver femur—type I î Cloverleaf skull—type II | Usually lethal |
| Osteogenesis imperfecta (OI) COL1A1 COL1A2 CRTAP/LEPRE1 | î Bone fractures î Irregularity and angulation to long bones î Decreased skull ossification î Irregular shape of ribs | Type II perinatal lethal Severity and features are variable based on the type of OI |
Collagen and Bone Disorders
Collagen/bone disorders can be diagnosed prenatally. The most common skeletal dysplasia disorders that are diagnosed prenatally are achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta (Table 12-6). Ultrasound findings include shortened limbs, 3 to 4 standard deviations below the mean for gestational age, as well as abnormalities in the skull, spine, and thorax. Options for further management of the pregnancy may depend on these ultrasound findings, as type II osteogenesis imperfecta and thanatophoric dysplasia are lethal.
SUGGESTED READINGS
Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. NEJM 2011 ;364(11 ):993-1004.
Allen LD, Sharland GK, Chita SK, et al. Chromosomal anomalies in fetal congenital heart disease. Ultrasound Obstet Gynecol 1991;1:8-11.
American College of Obstetricians and Gynecologists Committee on Practice Bulletins. ACOG practice bulletin no. 77: screening for chromosomal abnormalities. Obstet Gynecol 2007;109(1):217-227.
Barboza JM, Dajani NK. Prenatal diagnosis of congenital cardiac anomalies: a practical approach using two basic views. Radiographics 2002;22:1125-1138.
Centers for Disease Control and Prevention. National Center on Birth Defects and Developmental Disabilities. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/ncbddd/index.html. Accessed November 13, 2014. Copel JA, Cullen M, Green JJ, et al. The frequency of aneuploidy in prenatally diagnosed congenital heart disease: an indication for fetal karyotyping. Am J Obstet Gynecol 1988; 158:409-413.
Cuckle HS, Malone FD, Wright D, et al. Contingent screening for Down syndrome—results from the FaSTER trial. Prenat Diagn 2008;28(2):89-94.
Cunningham FG, Leveno K, Bloom SL, et al. Fetal imaging. In Cunningham FG, Leveno K, Bloom SL, et al, eds. Williams Obstetrics, 23rd ed. New York, NY: McGraw-Hill, 2010:349-373.
Cunningham FG, Leveno K, Bloom SL, et al. Prenatal diagnosis and fetal therapy. In Cunningham FG, Leveno K, Bloom SL, et al, eds. Williams Obstetrics, 23rd ed. New York, NY: McGraw-Hill, 2010:287-311.
Gabbe SG, Niebyl JR, Simpson JL, et al. Prenatal genetic diagnosis. In Gabbe SG, Niebyl JR, Simpson JL, et al, eds. Obstetrics: Normal and Problem Pregnancies, 6th ed. Philadelphia, PA: Saunders, 2012:210-236.
Laurence KM, James N, Miller MH, et al. Double blind randomized controlled trial of folate treatment before conception to prevent recurrence of neural tube defects. JAMA 1988; 260:3141-3145.
Leschot NJ, Verjaal M, Treffers PE. Risks of midtrimester amniocentesis: assessment in 3000 pregnancies. Br J Obstet Gynaecol 1985;92:804-807.
Malone FD, Canick JA, Ball RH, et al; First- and Second-Trimester Evaluation of Risk Research Consortium. First-trimester or second-trimester screening, or both, for Down's syndrome. N Engl J Med 2005;353:2001- 2011.
Nyberg DA, Souter VL, El-Bastawissi A, et al. Isolated sonographic markers for detection of fetal Down syndrome in the second trimester. J Ultrasound Med 2001;20:1053-1063.
Rhoads GG, Jackson LG, Schlesselman SE, et al. The safety and efficacy of chorionic villus sampling for early prenatal diagnosis of cytogenetic abnormalities. N Engl J Med 1989; 320:609-617.
Taipale P, Hiilesmaa V, Salonen R, et al. Increased nuchal translucency as a marker for fetal chromosomal defects. N Engl J Med 1997;337:1654-1658.