Corpus uteri
Endometrial hyperplasia
Endometrial hyperplasia forms a morphological continuum of abnormal proliferation ranging from focal glandular crowding or simple hyperplasia to well-differentiated adenocarcinoma.
Pathology
The 2014 World Health Organization (WHO) scheme distinguishes only two categories of endometrial hyperplasia: (1) hyperplasia without atypia; and (2) atypical hyperplasia/endometrial intraepithelial neoplasia (EIN) (1, 2).
Hyperplasia without atypia
This is an exaggerated proliferation of glands of irregular size and shape with increase in the gland-to-stroma ratio compared with proliferative endometrium, but without significant nuclear atypia. Hyperplasia without atypia is the result of unopposed oestrogenic stimulation. Progression to endometrial carcinoma occurs in 1-3% of women with hyperplasia without atypia
Atypical hyperplasia/endometrial intraepithelial neoplasia
This lesion shows marked glandular crowding, often as back-to- back glands, with little intervening stroma and cytological atypia. Epithelial cell nuclei are large and hyperchromatic with prominent nucleoli. One-quarter to one-third of these women will be diagnosed with endometrioid carcinoma at immediate hysterectomy or during the first year of follow-up. (2).
EIN refers to a monoclonal neoplastic growth of genetically altered cells with a greatly increased risk of becoming the endometrioid type of endometrial adenocarcinoma. The main diagnostic criterion of EIN is that the gland area exceeds that of the stroma (volume percentage stroma <55%). Atypical hyperplasia/EIN contains many of the genetic changes seen in endometrioid endometrial carcinoma, that is, microsatellite instability, and PTEN, KRAS, and CTNNBl (beta-catenin) mutation (1, 2).
Clinical features
Hysterectomy is usually the therapy of choice if a woman does not want more children.
Women who want more children or those with high operative risks may be treated with progestins.Endometrial adenocarcinoma
Endometrial carcinoma is the sixth most frequent cancer diagnosed in women globally with an age-standardized incidence rate of 8.2 per 100,000. It is the fourth most common cancer in women in industrialized countries and the most common gynaecological cancer. Three-quarters of women with endometrial cancer are postmenopausal. The median age at diagnosis is 63 years (1, 2).
Endometrial carcinoma is classified into two different types (Figure 70.5 and Table 70.1). Type I tumours (Figure 70.5a) (about 80%), endometrioid carcinomas, are often preceded by endometrial hyperplasia or EIN and are associated with oestrogenic stimulation. They occur mainly in pre- or perimenopausal women and are associated with obesity, hyperlipidaemia, anovulation, infertility, and late menopause. Typically, most endometrioid carcinomas are confined to the uterus and follow a favourable course. In contrast, type II tumours (Figure 70.5b) (about 10%) are non-endometrioid, largely serous carcinomas, arising occasionally in endometrial polyps. Type II tumours are not associated with oestrogen stimulation or hyperplasia, readily invade myometrium and vascular spaces, and are highly lethal (1).
Endometrial cancer is the most common extracolonic cancer in women with hereditary non-polyposis colon cancer syndrome, a defect in DNA mismatch repair that is also associated with breast and ovarian cancers (5).
Molecular pathogenesis
A dualistic model of endometrial carcinogenesis has been proposed. According to this model, normal endometrial cells transform into endometrioid carcinoma through replication errors, so-called microsatellite instability (Figure 70.5e) and subsequent accumulation of mutations in oncogenes and tumour suppressor genes. For non-endometrioid carcinomas, alterations of p53 (Figures 70.5f and 70.5g) and loss of heterozygosity on several chromosomes drive malignant transformation (5).
Five main molecular alterations have been described in type I endometrioid carcinomas: microsatellite instability (25-30% of cases) (Figure 70.5e); PTEN mutations (30-60%); PIK3CA mutations (26-39%); ARIDlA mutations (20%); K-RAS mutations (1030%); and CTNNBl (beta-catenin) mutations with nuclear protein accumulation (25-38%). In contrast, most type II non- endometrioid carcinomas have p53 mutations (Figure 70.5g), Her-2/neu amplification, and loss of heterozygosity on several chromosomes. Non- endometrioid carcinomas may also derive from endometrioid carcinoma with microsatellite instability through tumour progression and subsequent p53 mutations (5).
The Cancer Genome Atlas (TCGA) has conducted the most comprehensive genomic analysis of endometrial carcinomas reported to date (8). TCGA has expanded the dualistic classification of endometrial carcinoma (types I and II) to four distinct molecular subgroups: (1) an ultramutated POLE subgroup; (2) a hypermutated microsatellite unstable subgroup; (3) a copy-number low/micro- satellite stable subgroup; and (4) and a copy-number high/serous- like subgroup. Even if overlapping of the molecular genetic findings makes it still difficult to separate significant prognostic categories, POLE mutations predict favourable prognosis, particularly in highgrade tumours. Patients with endometrioid tumours that are serous- like at the molecular level might benefit from treatments that are typically used for serous carcinomas (6).
Pathology
Endometrioid adenocarcinoma of the endometrium
This type of endometrial cancer is composed entirely of glandular cells and is the most common histological variant (80-85%) (Figure 70.5c). The FIGO system divides this tumour into three grades on the basis of the ratio of glandular to solid elements, the latter signifying poorer differentiation. Less common histological variants include endometrioid adenocarcinoma with squamous differentiation and the mucinous and secretory types, both associated with good prognosis (1-3).
Non- endometrioid endometrial carcinomas
They are aggressive as a group, and histological grading is not clinically useful, all cases being considered high grade:
• Serous adenocarcinoma histologically resembles, and behaves like, high-grade serous adenocarcinoma of the ovary (Figure 70.5d). It often shows transtubal spread to peritoneal surfaces. An intraepithelial form has been termed ‘serous endometrial intraepithelial carcinoma' (serous EIC), not to be confused with EIN, described earlier. Patients with this type of tumour need to be staged and treated as if they had ovarian cancer.
• Clear cell adenocarcinoma is a tumour of older women. It contains large cells with abundant cytoplasmic glycogen (‘clear cells')
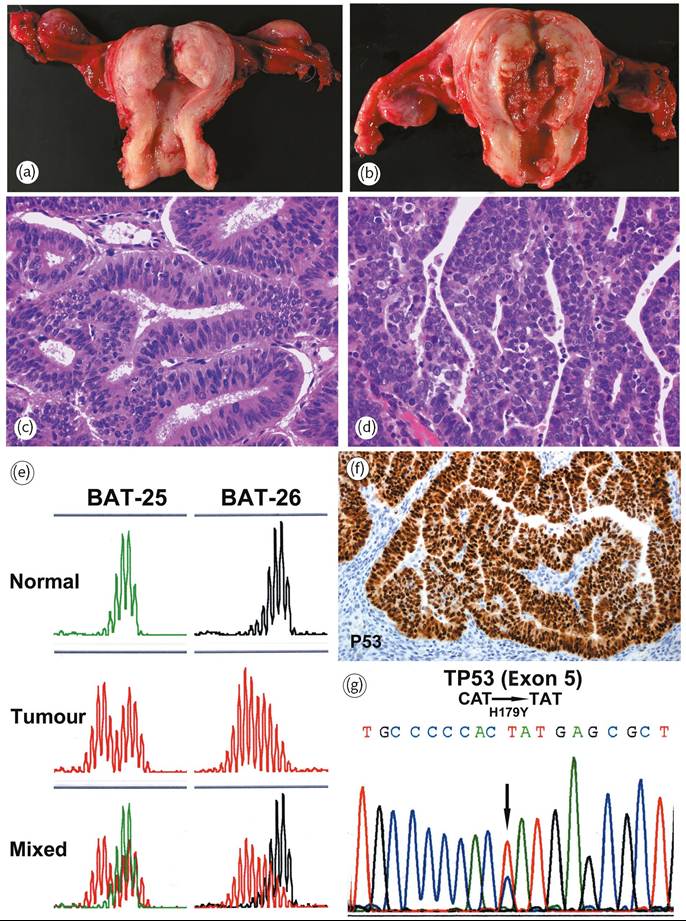
Figure 70.5 Adenocarcinoma of the endometrium. (a) Endometrioid carcinoma (type I). Polypoid endometrial tumour with only superficial myometrial invasion. (b) Serous (non-endometrioid) carcinoma (type II). Large haemorrhagic and necrotic tumour with deeper myometrial invasion. (c) Well-differentiated (grade 1) endometrioid adenocarcinoma. The neoplastic glands resemble normal endometrial glands. (d) Serous (non- endometrioid) carcinoma exhibiting stratification of markedly atypical tumour cells with numerous mitoses. (e) Endometrioid carcinoma. MLH1 inactivation by promoter hypermethylation is the most common cause of the microsatellite instability (MI) phenotype in endometrioid endometrial carcinoma. Progressive accumulation of alterations secondary to MI affects important regulatory genes, and promotes carcinogenesis. (f) Serous (non- endometrioid) carcinoma usually shows a strong p53 overexpression as a result of TP53 mutation (g).
or cells with bulbous nuclei that line glandular lumina (‘hobnail cells’). Clear cell carcinomas have poor prognosis.
• Carcinosarcoma (malignant mixed mesodermal tumour): in this highly malignant tumour, pleomorphic epithelial cells intermingle with areas showing mesenchymal differentiation.
These mixed neoplasms are derived from a common clone thought to be of epithelial origin. Overall 5-year survival is 25% (1, 2).Clinical features
Unlike cervical cancer, endometrial cancer may spread directly to para-aortic lymph nodes, thereby skipping pelvic nodes. Patients with advanced cancers may also develop pulmonary metastases (40% of cases with metastases).
Women with well-differentiated cancers confined to the endometrium are usually treated by simple hysterectomy. Postoperative radiation is considered if (1) the tumour is poorly differentiated or non-endometrioid in type; (2) the myometrium is deeply invaded; (3) the cervix is involved; or (4) the lymph nodes contain metastases.
Survival in endometrial carcinoma is related to multiple factors: (1) stage, histotype, and, for endometrioid tumours, grade; (2) age; and (3) other risk factors, such as progesterone receptor activity, depth of myometrial invasion, and extent of lymphovascular
Table 70.1 Clinicopathological features of endometrial carcinoma
| Type I: endometrioid carcinoma | Type II: serous carcinoma | |
| Age | Pre- and perimenopausal | Postmenopausal |
| Unopposed oestrogen | Present | Absent |
| Hyperplasia precursor | Present | Absent |
| Grade | Low | High |
| Myometrial invasion | Superficial | Deep |
| Growth behaviour | Stable | Progressive |
| Genetic alterations | Microsatellite instability, PTEN, PIK3CA, β-catenin | TP53 mutations, loss of heterozygosity |
invasion (7).
Actuarial survival of all patients with endometrial cancer following treatment is 80% after 2 years, decreasing to 65% after 10 years. Tumours that have penetrated the myometrium or invaded lymphatics are more likely to have spread beyond the uterus. Endometrial cancers involving the cervix have a poorer prognosis. Spread outside the uterus entails the worst outlook (3, 7).Endometrial sarcomas
Currently, endometrial sarcomas are classified into three categories: (a) low-grade endometrial stromal sarcoma (LGESS); (b) high-grade endometrial stromal sarcoma (HGESS); and (c) undifferentiated endometrial sarcoma (UES) (2). LGESSs represent less than 2% of uterine cancers. They may be polypoid or may diffusely invade the myometrium. The tumour cells resemble endometrial stromal cells in the proliferative phase. Nuclear atypia may be minimal to severe and mitotic activity may be restrained. Expression of CD10 and oestrogen and progesterone receptors helps confirm the diagnosis. The most common cytogenetic abnormality of LGESS is a recurrent translocation involving chromosomes 7 and 17 t(7;17) (p15;q21) which results in a fusion between JAZF1 and SUZ12 genes (formerly designated as JJAZ1) (1, 2).
The recently re-established HGESS has features intermediate between LGESSs and undifferentiated sarcomas. It may appear as intracavitary polypoid or a mural mass. Microscopically, it consists predominantly of high- grade round- cells which are sometimes associated with a low-grade spindle cell component usually fibromyxoid. Mitotic activity is very striking and typically greater than ten per 10 high power fields (HPF). Necrosis is usually present. HGESS typically harbours the YWHAE-FAM22 genetic fusion as a result of t(10;17)(q22;p13) (1, 2).
Higher-grade poorly differentiated sarcomas originating in the endometrium are designated as undifferentiated endometrial sarcoma (1, 2).
Clinical features
Many years may elapse before LGESSs recur clinically, and metastases may occur even if the original tumour was confined to the uterus at initial surgery. Recurrences usually involve the pelvis first, followed by lung metastases. Prolonged survival and even cure are feasible, despite metastases. By contrast, UESs recur early, generally with widespread metastases. In comparison to patients with LGESSs, those with HGESSs and UES, have earlier and more frequent recurrences (often <1 year) and are more likely to die of disease. LGESSs can be successfully treated with surgery and progestin therapy, with an expectation of 90% survival 10 years after diagnosis (1, 2).
Uterine adenosarcoma
Uterine (Mullerian) adenosarcoma is a distinctive low-grade tumour with benign glandular epithelium and malignant stroma. It should be distinguished from carcinosarcoma, in which both epithelial and stromal elements are malignant and which is highly aggressive. One-quarter of patients with adenosarcoma, particularly cases with myometrial invasion and sarcomatous overgrowth, eventually succumb to local recurrence or metastatic spread (1, 2).
Leiomyosarcoma
Leiomyosarcoma is a malignancy of smooth muscle origin whose incidence is only 1/1000 that of leiomyoma. It accounts for 2% of uterine malignancies. Its pathogenesis is uncertain. Women with leiomyosarcomas are on average more than a decade older (age >50 years) than those with leiomyomas, and the malignant tumours are larger (10-15 cm vs 3-5 cm) (1, 2).
Pathology
Leiomyosarcoma should be suspected if an apparent leiomyoma is soft, shows areas of necrosis on gross examination, or has irregular borders (invasion of adjacent myometrium). Mitotic activity (ten or more mitoses per 10 HPFs), nuclear atypia, and geographic necrosis are the best diagnostic criteria (Figures 70.6a and 70.6b). Myxoid and epithelioid leiomyosarcomas may contain only five mitoses per
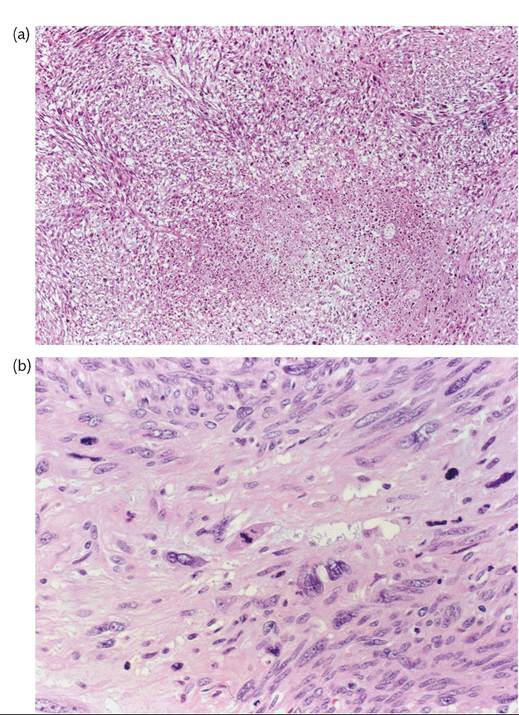
Figure 70.6 Leiomyosarcoma of the uterus. (a) A zone of coagulative tumour necrosis appears demarcated from the viable tumour. (b) The tumour shows considerable nuclear atypia and abundant mitotic activity.