Oncogenetics
Principles
‘All cancer is genetic but some cancers are more genetic than others.’ This paraphrase from George Orwell’s Animal Farm conveys the essence of the genetic basis of malignancy.
At the tissue level, all cancers are the result of the cumulative effect of environmental insults and genetic changes leading to dysregulated, uncontrolled growth of abnormal clones of cells. In a minority of cases, these tissue-specific genetic alterations leading to cancer are predisposed to by highly penetrant, inherited germline mutations (122). Sporadic cancers are the result of detrimental genetic dysregulation of growth in the affected organ whereas in the so-called inherited cancers, the underlying factors are genetic changes affecting all cells of the body. In reality, there exists a spectrum between true sporadic and ‘inherited’ disease with many cancers being the result of the cumulative effect of a number of lower-penetrance familial genetic alterations (123-126).Genes in which mutations predispose to cancer are broadly classified under two categories: proto-oncogenes and tumour suppressor genes (TSGs). Genes of both categories fulfil important functions in the human body. Proto-oncogenes play important roles in regulated growth and proliferation and include growth factors and components of signal transduction pathways. Mutations can result in a proto-oncogene becoming an oncogene, driving uncontrolled growth and tumour formation. On the other hand, TSGs function as guards against unregulated growth either by controlling proliferation, promoting programmed cell death (apoptosis) of abnormally proliferating cell clones, or, at a more fundamental level, by repairing damage to DNA. This class of TSGs are known as DNA repair genes and mutations in them do not directly lead to tumori- genesis but increase the likelihood of mutations in other TSGs or proto-oncogenes.
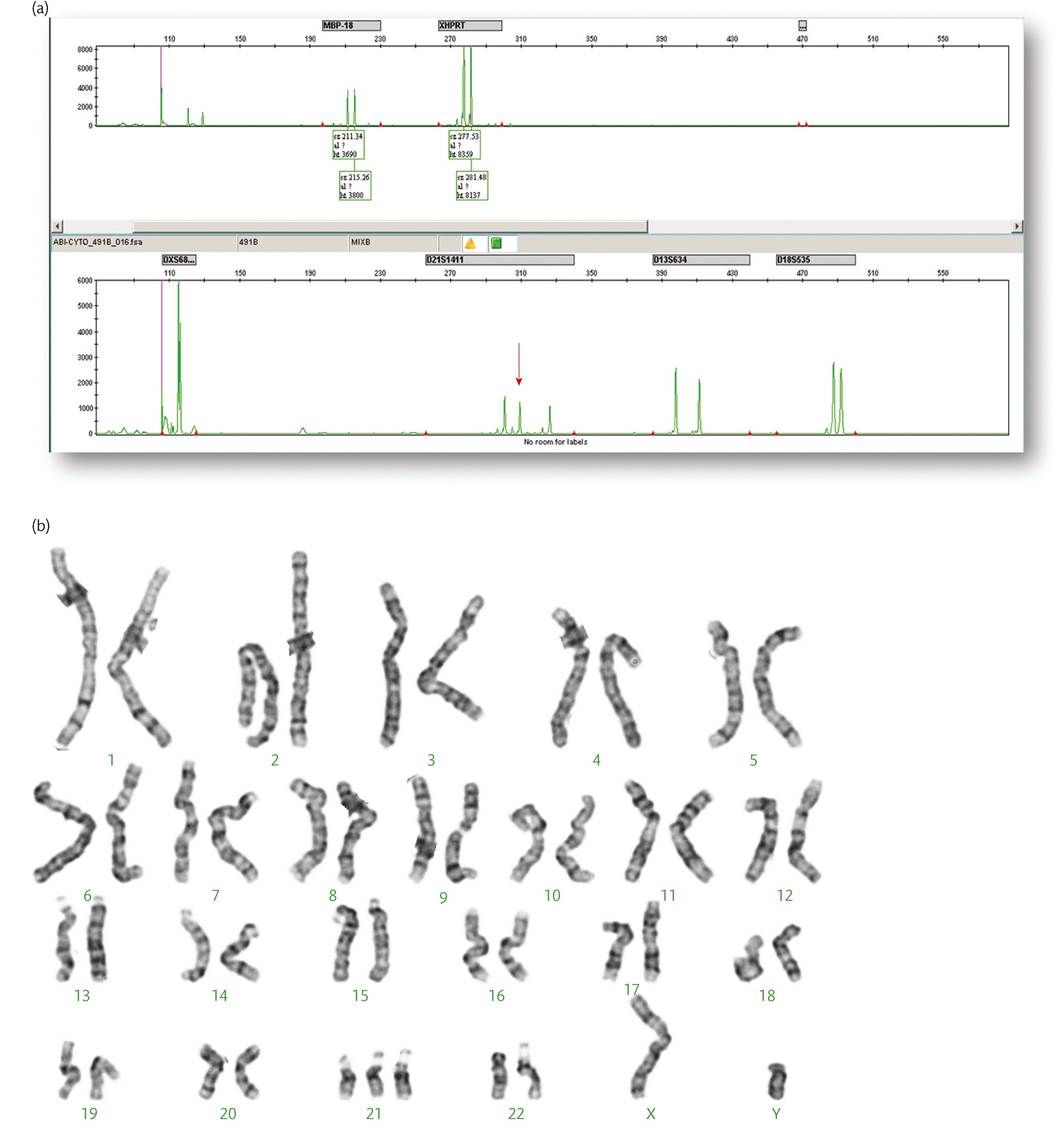
Figure 4.7 Quantitative fluorescent PCR electropherogram in (a) showing three copies of amplified markers specific for chromosome 21 as indicated by the three peaks (arrow). Trisomy 21 is confirmed by the G-banded karyotype in (b).
Courtesy Dr Carolina Sismani, Cyprus Institute of Neurology & Genetics.
Mutations in both proto-oncogenes and TSGs are linked to specific cancer predisposition syndromes which are most commonly inherited in an autosomal dominant manner. In the case of protooncogenes, gain-of-f unction mutations in the heterozygous state confer increased cancer susceptibility as the abnormal gene products of the mutated allele drive growth. On the other hand, both alleles of TSGs need to be altered at the tissue level for that specific tumour suppressor function to be deficient. So, the TSG mutations (and the resulting syndromes) are inherited in the heterozygous state but the inactivation of the second allele at the organ level is required for the sequence of events leading to tumour formation to be initiated. This is the principle underlying the so-called Knudson's two-hit hypothesis (one germline and one tissue-specific) (127). In essence, TSG mutations are inherited dominantly but act recessively at the level of the organs.
Genetics and gynaecological malignancies
Though the majority of gynaecological malignancies occur sporadically, both ovarian and endometrial cancers are prominent features of key cancer predisposition syndromes, illustrating the role of germline mutations in the aetiology of cancer. The lifetime risk of women developing ovarian cancer is very significantly increased in two of the most common cancer predisposition syndromes: BRCA hereditary breast and ovarian cancer syndrome and Lynch syndrome. Both syndromes are caused by mutations in TSGs and in both cases these are TSGs involved in DNA repair.
The BRCA hereditary breast and ovarian cancer syndrome is caused by mutations in the genes BRCA1 and BRCA2 which are involved in post-replication DNA repair (128-130).
The gene products repair breaks following DNA replication and loss of this repair function leaves the cells exposed to the detrimental effects of uncorrected errors. It is the accumulation of such errors that eventually leads to tumorigenesis. The presence of BRCA mutations in the heterozygous state is not sufficient to disrupt the DNA repair ability of the cell. It is the loss of the functioning second allele that occurs through an independent event (second hit) at the tissue level that is the critical step. The syndrome is associated with a significant increase in breast and ovarian cancer risks as well as the risk of other malignancies including prostate and pancreas (131-134).Lynch syndrome is caused by mutations in a family of genes involved in DNA repair known as mismatch repair genes (135-138). The function of the gene products is to correct errors in basematching following DNA replication. As with BRCA, the presence of germline mutations in these genes confers susceptibility to cancer and it is the loss of the second, functioning allele that results in loss of the mismatch repair function and the subsequent sequence of events that leads to cancer development. The syndrome is associated with an increased risk of many cancers including colorectal, endometrial, gastric, and ovarian (139-141).
Oncogenetics in clinical practice
In assessing a woman diagnosed with a malignancy such as ovarian or endometrial cancer, a gynaecologist may consider the possibility that the disease developed in the context of a cancer predisposition syndrome. The woman's age, previous personal history of cancer, and relevant family history of malignant disease can be pointers to a possible genetic predisposition and the genogram is particularly useful in that context. A woman being diagnosed with ovarian cancer in her early 40s with a strong family history of breast cancer will need to be counselled for the possibility of an underlying BRCA mutation. In a woman diagnosed with endometrial cancer in her 50s who has a past history of colorectal cancer, the possibility of Lynch syndrome will need to be considered.
Usually, a referral will be made to the clinical genetics service for consideration of genetic testing.The identification of an underlying genetic predisposition is important and may influence the therapeutic options available and subsequent surveillance. For example, women with breast cancer and an underlying BRCA mutation have a significant lifetime risk of developing a second primary breast cancer (142-145). This has an impact on the choice of surgical procedure and follow-up and may also raise the possibility of prophylactic surgery (contralateral mastectomy) to manage the risk (146-149). Women may also consider prophylactic bilateral salpingo-oophorectomy after completing their families to reduce the significant associated ovarian cancer risk as well as the breast cancer risk in the premenopausal period, though the latter is currently the subject of debate (150-153). In Lynch syndrome, the option of prophylactic hysterectomy and bilateral salpingo-oophorectomy should be discussed (154, 155).
A test for a cancer predisposition syndrome offered to a woman already affected from cancer is described as a diagnostic test. Tests offered to unaffected family members to establish whether they carry a known familial mutation are described as predictive. Women offered genetic testing should be counselled comprehensively about the nature of the tests and the implications of their results. Such counselling should address the complex medical, ethical, and psychosocial aspects that are inherent to this process (156-159). In diagnostic tests, the possibility of identifying variants of unknown significance should be discussed (160, 161). Beyond the relevance to management decisions, the psychological impact, at a time when a woman is dealing with her own diagnosis of cancer, should be explored, also taking into account the implications for other family members who are potentially at risk.
There are a number of considerations in relation to predictive tests. The ability to achieve prevention through close surveillance is one of the main indications and this relies on the availability of validated screening programmes for the relevant cancers.
Examples include colonoscopy screening for Lynch syndrome (162, 163) and breast screening with magnetic resonance imaging in women with BRCA mutations (164, 165). Unaffected women also have the option of prophylactic surgery and the possibility of modifying risks through changes in lifestyle and avoidance of additional risk factors should also be explored.The inheritance implications for existing or future children should be discussed. Some women may consider the option of prenatal (or preimplantation) genetic diagnosis though this is unusual because of the incomplete penetrance of many of the syndromes and the opportunities for prevention (166, 167). The potential impact on the ability of women to secure appropriate personal insurance once tested positive for a cancer predisposition syndrome needs to be part of pretest counselling though legislation has been passed in many countries to protect individuals from resulting discrimination (168, 169).
Novel therapeutic implications of oncogenetics
Establishing the molecular basis of cancer susceptibility has revealed new potential targets for treatment. Paradoxically, the chink in the armour of the cell's repair mechanisms may offer hope for novel therapeutic targeting of tumour cells (170). As described previously, loss of BRCA DNA-repair activity leaves cells vulnerable to other mutations and predisposes to oncogenesis. If manipulated, however, that disturbance in DNA repair ability can compromise the viability of tumour cells leading to cell death. One such manipulation is the use of poly ADP ribose polymerase (PARP) inhibitors which makes cells even more dependent on BRCA and has shown promising results in treating tumours which form part of the BRCA hereditary breast and ovarian cancer syndrome. PARP supports single- stranded DNA repair and if its function is impaired, cells rely on more complex double-stranded repair mechanisms, such as homologous recombination and non-homologous end joining which are mediated by BRCA. If BRCA-mediated repair is itself deficient as a result of a germline mutation then PARP inhibition tips the balance towards genomic instability and cell death in these tumour cells (171-174).