Genetic aspects of the clinical encounter
The assessment of the inheritance pattern of disorders and traits is by no means restricted to the work of clinical geneticists and a family history is an essential component of most clinical encounters.
The relevant information is depicted in the genogram which can be updated as and when necessary and which underpins genetic interpretation. A number of computer programs allow genograms to be generated and updated electronically. However, in the authors experience, a well-constructed hand-written genogram is an efficient and effective way of recording this information. It conveys details on diagnoses, causes of death, and adverse pregnancy outcomes and is an accurate record of genetic relationships. Certain basic rules underlie reliable and reproducible genogram generation. It should record the date when first constructed as well as the dates of any updates, full names of affected family members and dates of birth (not ages as this will hinder interpretation), and a diagnostic index and should be based on the consistent use of established symbols (Figure 4.6).The genogram helps establish modes of inheritance for genetic conditions, demonstrates risk patterns, and identifies clusters of events (such as recurrent early pregnancy loss) that are relevant to the consultation. In a woman with a new diagnosis of ovarian cancer, a careful record should be made of other cases of cancer, clustering of cases on either of the two sides of the woman's family, and the age at diagnosis. In addition to other cases of ovarian malignancy, particular attention is paid to cancers that point towards the possibility of an underlying cancer predisposition syndrome (breast, endometrial, colorectal).
The preconception interview
Genetic concepts frequently feature in clinical consultations and women (or couples) request counselling when planning to start or
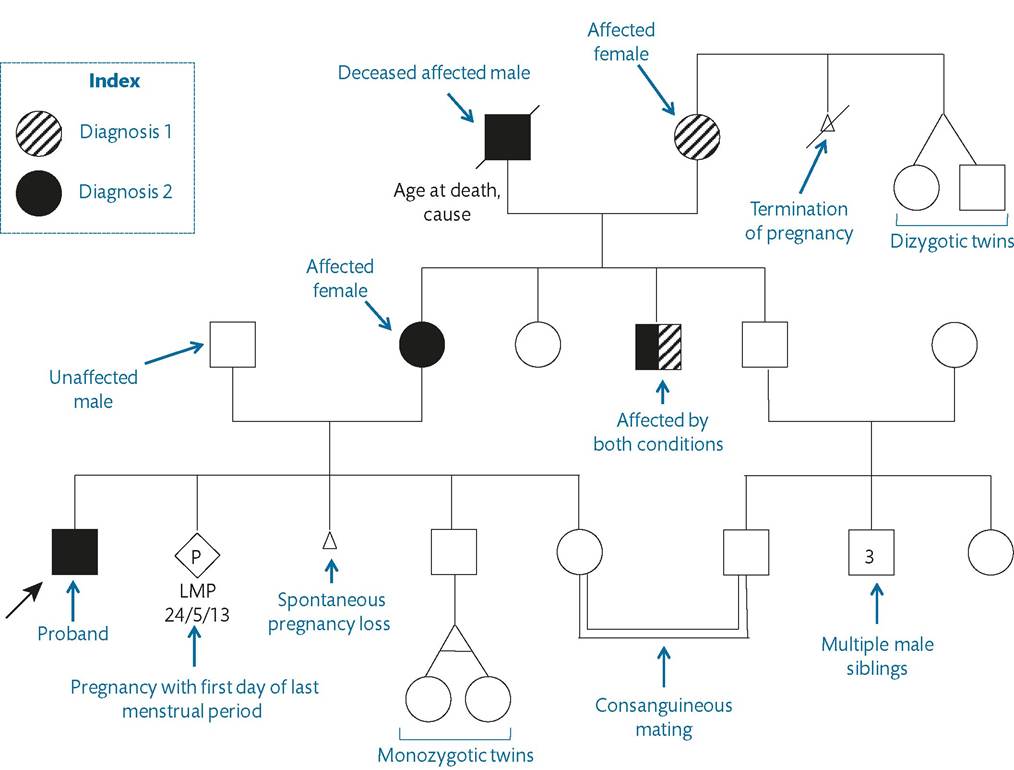
Figure 4.6 Symbols used in the construction of a genogram in conjunction with relevant information.
Sample genogram drawn to illustrate use of the symbols.extend their family or in an established pregnancy. Issues that feature in the preconception interview include infertility or subfertility, health-promotion advice, the impact of pregnancy on a pre-existing medical condition, and possible genetic risks to the baby. Discussion of most of these issues is beyond the scope of this chapter and the following subsections will concentrate on the approach to possible genetic implications in a future pregnancy.
The prospective parents may themselves be affected or concerns may be raised because of a previously affected child, a possible or confirmed genetic diagnosis in the family, a genetic condition that is prevalent in the specific population, the possible implications of consanguinity, and repeated losses of pregnancy or birth of babies with congenital malformations in the family. Whatever the specific concerns, a detailed family history should be taken and a genogram drawn capturing as many relevant details as possible. This will form the basis for subsequent discussion, planning, investigation, and relevant counselling.
Affected prospective parents
A distinction should be made between monogenic conditions, conditions with multifactorial aetiology and recognized genetic predisposition, and sporadic disease. The rest of the discussion will concentrate on the scenario of prospective parents with single gene disorders.
A woman with a diagnosis of neurofibromatosis type 1 (NF1) needs to be counselled that, as this is an autosomal dominant condition, the baby in a future pregnancy will have a one in two chance of inheriting the responsible mutation in the NF1 gene. Logical follow-up questions include whether inheritance of the mutation means that the child will develop the condition and if the child will be affected in the same way as the parent. These are key questions in genetics and relate to the concepts of penetrance and expressivity.
Penetrance is the percentage of individuals carrying a pathogenic dominant mutation known to cause a condition who will go on to develop the condition regardless of its severity.
Expressivity describes the variation in severity of a genetic condition between affected individuals who carry the same mutation (parent and child, for example). NF1 is virtually fully penetrant and a child inheriting an NF1 mutation is expected to develop the condition. On the other hand, this is a condition with very variable expressivity and a mildly affected parent with mostly cutaneous manifestations may have an affected child with complex plexiform neurofibromas or an optic glioma. In contrast, a parent affected by achondroplasia, another fully penetrant autosomal dominant condition, can be counselled that an affected child will have the clinical features of the condition including severe short stature as the variability in expressivity seen in NF1 does not apply. These considerations will have to be applied to the individual conditions.Another phenomenon relevant to some autosomal dominant conditions is that of anticipation. It describes an increase in severity from generation to generation and is characteristically observed in disorders caused by a type of insertion mutation known as triplet repeat expansion. Instability of the number of triplet repeats during gametogenesis underlies this phenomenon which in some conditions is largely dependent on the sex of the transmitting parent. For example, in Huntington disease, large expansions typically occur in the paternal line (51).
Autosomal recessive conditions develop when both alleles (maternally and paternally inherited) carry pathogenic mutations and are exemplified by affected children born to unaffected carrier parents. In the case of a woman affected by beta thalassaemia requesting counselling about a future pregnancy, the main determinant is the carrier status of her partner. Whereas the affected woman is certain to pass on one of the two mutated alleles to her offspring, only if the partner is a carrier will there be a chance of the children being affected (a one in two chance for each pregnancy).
With a non-carrier partner, there is certainty of the children being unaffected carriers.In X-linked recessive conditions, such as X-linked hypohidrotic ectodermal dysplasia, affected fathers cannot pass the mutation to their sons but are certain to pass it on to their daughters who are obligate carriers and may exhibit mild clinical features. These specific considerations do not apply to disorders such as Duchenne muscular dystrophy where affected males do not reproduce because of the severity of the condition.
Whatever the mode of inheritance, there are certain key considerations at the preconception stage. Has the condition been diagnosed based only on clinical criteria or has it been confirmed by genetic testing? Identification of parental mutations is crucial to the planning for preimplantation genetic or prenatal diagnosis and a referral to the clinical genetics service at this stage is appropriate. Moreover, in the absence of genetic confirmation, the possibility of using antenatal sonographic markers needs to be explored. Examples include sonographic evaluation of fetuses for fractures or deformities in osteogenesis imperfecta or the tumours related to tuberous sclerosis. These features, however, are variable and mainly detectable in the latter stages of pregnancy (52-54).
Previous affected child
Providing counselling on recurrence risks is a frequent component of preconception consultations and a key task for clinical geneticists (55, 56). First, it must be established whether the affected child has a genetic diagnosis following a single gene inheritance pattern and whether laboratory confirmation has taken place.
A basic principle is that the recurrence risk depends on the genetic status of the parents. In autosomal dominant disorders, a pathogenic mutation in an affected child has either been inherited from one of the parents or has arisen as a new mutation (de novo) in the child. This will vary depending on the specific condition. In NF1, about 50% of cases are the result of de novo mutations and in achondroplasia about 80% of affected children are born to unaffected parents.
Depending on the condition, the parental status can be established either on the basis of clinical features or by genetic testing, provided the child's mutation has been identified. The clinical assessment will be tailored to the individual condition and will comprise careful examination and appropriate referrals and investigations.If one of the parents is affected, the recurrence risk is one in two as described in the previous section. If the parents are unaffected, then the recurrence risk is low but may be higher than the population risk for the condition. This could be accounted for by the possibility of germline mosaicism which implies that the mutation is present in a subset of gametes in one of the parents who is clinically unaffected. In tuberous sclerosis, for example, this risk is estimated to be about 1-2% and different figures apply to different conditions (57).
When the affected child has an autosomal recessive condition, such as beta thalassaemia or cystic fibrosis, it can be safely assumed that both mutations have been inherited from the parents since, with very rare exceptions, the rate of de novo recessive mutations is extremely low. When both parents are carriers, the recurrence risk is one in four.
In X-linked recessive disorders, the recurrence risk depends on the genetic status of the mother. In Duchenne muscular dystrophy, a mother who is a carrier for a DMD mutation has a one in four recurrence risk (one in two if the baby is known to be a boy). The mother can be tested for the mutation or the family history may confirm her as an obligate carrier if there is another affected relative in the maternal line. If the mother is shown not to be a mutation carrier, the affected child either has a de novo mutation or the mother has germline mosaicism.
When dealing with conditions that do not follow Mendelian inheritance, such as many isolated congenital malformations, empiric risks based on observed recurrence data can be given. A comprehensive assessment of the affected child to rule out possible syndromic associations is necessary before such empiric risks are used.
There are also occasions when it may not be possible to confidently distinguish between an undiagnosed single gene disorder that is recessively inherited and one that is the result of a de novo dominant mutation. Recurrence risks differ greatly in the two scenarios and clinical geneticists may use intermediate risk estimates based on the level of probability in the specific clinical context. If a recurrence does occur or if genetic testing subsequently confirms a single gene disorder, future advice will be adjusted accordingly.Familial conditions
Women and their partners who are neither affected nor have affected children may raise the possibility of a genetic condition running in the family. The overall objective is to establish the likelihood of unaffected prospective parents being carriers for a genetic condition so that appropriate genetic testing can be offered or suggest that such testing may not be indicated.
Constructing a genogram is instrumental in helping the clinician understand and quantify any possible risks to a future pregnancy and counsel appropriately. The information needs to be as accurate and as comprehensive as possible and women are often asked to return to the clinic with additional details and clarifications in order to fill in any gaps and it may be necessary for consent to be obtained from family members in order to access details of a specific diagnosis.
When interpreting the genogram, the ratio of affected males to females, the severity in males compared to females, and any male-t o-male transmission need to be assessed in evaluating the
possibility of an X-linked disorder. Successive generations being affected may point towards autosomal dominant inheritance and the skipping of generations raises the possibility of reduced penetrance. Consanguinity supports autosomal recessive conditions and the lack of any transmission from males would be consistent with mitochondrial (non-Mendelian) inheritance. These considerations form the basis of genogram analysis which should be used in the context of the available clinical details. This interpretation may be hampered by the small number of affected individuals in a family. Moreover, there may be insufficient evidence to suggest a single gene disorder.
Risk in the absence of a familial diagnosis
It is also possible for women or couples to request genetic counselling even if there are no documented cases of possible genetic disorders in the family. A careful family history should be taken as this may reveal potentially relevant diagnoses. Counselling may be requested on the basis of parental consanguinity or the prevalence of a genetic disorder in a population.
If the prospective parents are related to each other, there is primarily an increased risk of the baby being affected by autosomal recessive conditions as a result of the parents sharing a proportion of their genome depending on the degree of relationship. It is estimated that humans carry about one to two harmful autosomal recessive alleles and consanguinity increases the chance that the same pathogenic allele will be inherited by a baby from both parents. Couples need to be counselled about this with the relevant level of estimated and empiric risk being conveyed (55, 58).
The couple's ethnic background may also identify genetic risks and targets for testing particularly in relation to the carrier state for autosomal recessive disorders. In Northern European populations, the carrier rate for cystic fibrosis is about 1 in 25 and it is possible that a couple may request carrier testing (59). The carrier rate for beta thalassaemia is very high in some Mediterranean countries (in Cyprus it is as high as one in seven) and it is very important that prospective parents are counselled appropriately and appropriate testing is offered (60).
Recurrent early pregnancy losses
About 15% of all recognized pregnancies miscarry and chromosomal abnormalities account for about half of these losses (61, 62). In recurrent miscarriage, affecting up to 5% of couples if defined as two or more losses, products of conception are analysed cytogenetically to establish a diagnosis and determine whether balanced rearrangements in the parents might be the underlying factor (62-64).
For example, women carrying a Robertsonian translocation involving chromosomes 14 and 21 have a 10-15% risk of having a baby with Down syndrome and preimplantation and prenatal options need to be discussed (65-67). When a parent is found to be a carrier of a balanced reciprocal translocation, the associated risk of a viable imbalance needs to be assessed and appropriate plans need to be made in a future pregnancy (68, 69).
Communication aspects of genetic counselling
One of the central pillars of genetic counselling is the element of non-directiveness. Clinical geneticists and other clinicians and professionals involved in genetic counselling have a responsibility to provide their patients with the necessary information in order for them to decide on their preferred course of action (55, 70, 71). What needs to be thoroughly explored is a plan that is appropriate for the individuals involved in a particular context and at a particular moment in time. This principle is illustrated well in the context of a prenatal consultation. This is a period during which decisions need to be made regarding invasive prenatal tests and often about whether to continue with a pregnancy or proceed with termination.
Given the inherent uncertainty and complexity of many of these issues, it is not surprising that many women request guidance from their doctor in order to make a choice. In the author's opinion, clinicians would be most helpful by carefully exploring the various options with their patients with well-supported and constructive arguments on the advantages, disadvantages, and limitations of the various courses of action, empowering them to make informed choices. Moreover, directiveness in counselling can be problematic in situations, for example, where there are differing approaches among couples themselves. A good review of these issues is provided by Harper (55).
Preimplantation genetic diagnosis
The evolution of assisted reproduction has transformed the prospects for many infertile and subfertile couples. It has also made possible genetic scrutiny of the preimplantation embryos before these are returned to the uterus based on biopsy at the cleavage or blastocyst stage (72-74). This approach offers significant advantages for couples who have been identified at the preconception consultation as being at increased risk of having babies with genetic conditions.
There are two main categories of preimplantation genetic scrutiny: preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS). PGD is used to select unaffected embryos where there is high genetic risk for a specific disorder. The desire not to have children affected by a specific condition coupled with the unwillingness to consider prenatal diagnosis and possible termination of an established pregnancy is one of the main indications for considering PGD. The two main examples are inherited chromosomal disorders and monogenic diseases. The diagnosis of chromosomal abnormalities is based on FISH (75, 76) and of single gene disorders on PCR techniques (73, 77). The laboratory must have a validated preimplantation protocol for the specific diagnoses.
PGS is used to screen and select embryos free from chromosomal aneuploidy or other chromosomal imbalance, aiming to increase the chances of a successful pregnancy and take-home baby rates (78). Indications for PGS include advancing maternal age, recurrent early pregnancy loss, recurrent implantation failure, and severe male factor infertility and in these cases the parents do not have a known genetic risk or chromosomal rearrangement (79-82). The usefulness of PGS in improving pregnancy outcomes is under debate and may be limited by early embryo mosaicism and limitations of FISH techniques (83, 84). The use of array CGH and nextgeneration sequencing techniques may improve outcomes in this context (85, 86).
Prenatal testing
The monitoring and testing of a pregnancy known to be at genetic risk is usually planned at the preconception stage. Depending on the circumstances, it may involve invasive or non-invasive genetic testing, sonographic assessment, or a combination of these.
Mutation testing is still most commonly performed through invasive techniques: either a chorionic villous sampling, usually performed between 11 and 13 weeks of gestation, which tests the genetic material of placental tissue of fetal origin, or amniocentesis, usually performed between 15 and 20 weeks of gestation, which allows the examination of the genetic material of fetal cells suspended in the amniotic fluid. Both techniques are associated with a small, but not insignificant, risk of pregnancy loss of 0.5-1% (87). Another form of invasive testing, percutaneous umbilical cord sampling, is not commonly performed for genetic diagnosis and its use is largely restricted to haematological assessment of the fetus suspected to have severe anaemia and thrombocytopenia (88).
The demonstration of free fetal DNA in the maternal plasma has been the basis for developing alternative, non-invasive approaches to prenatal diagnosis (89, 90). An early application was the use of this free fetal DNA for the determination of the sex of the fetus. This is very important for pregnancies at risk of X-linked recessive disorders such as haemophilia A or Duchenne muscular dystrophy. As female fetuses are at most at risk of being carriers, this reduces the need for invasive procedures. The test, which is commonly performed after the eighth week of gestation, is based on the identification of male sex-determining region Y (SRY) sequences in the maternal plasma and the use of real-time PCR is associated with very high sensitivity and specificity (91-94). A fetal sonographic technique has also been developed which assesses the angle between the genital tubercle and the lumbosacral vertebral line (95). This is only feasible, however, after 11 weeks and its accuracy improves with increasing gestation (96). Some centres also advocate a combination of the two techniques (92, 97).
Non-invasive testing of free fetal DNA has also been used in the prenatal diagnosis of paternally inherited or de novo autosomal dominant conditions where mutations detected in the maternal plasma must be of fetal origin (98, 99). Moreover, it can be used to exclude inheritance of the paternal mutated allele in autosomal recessive cases where this is different from the maternal mutation (100, 101). The early techniques have been based on the use of PCR which requires prior knowledge of the mutation to be tested and its use in de novo diagnoses is limited to conditions such as achondroplasia where the vast majority of cases are caused by a single mutation. There is evidence that the application of next-generation sequencing techniques may improve the scope and accuracy of such testing in the future (102).
In pregnancies with no specific genetic risks identified in the preconception period, antenatal screening may identify risk factors for genetic diagnoses. Established aneuploidy screening programmes combine biochemical testing with sonographic markers to modify the age-related risk (103). When an invasive test is indicated in high-risk pregnancies, many laboratories use rapid an- euploidy testing such as interphase FISH (104, 105) or quantitative fluorescent PCR (106, 107) (Figure 4.7). There is a debate about the cost-effectiveness and additional diagnostic yield of routinely proceeding with a karyotype if these rapid tests are negative (108, 109). However, individual cases need to be evaluated based on the indications for the invasive test (such as sonographic abnormalities) and the possibility of missing other chromosomal abnormalities should be taken into account.
Recently, testing for fetal aneuploidy has been revolutionized by the introduction of non-invasive techniques using massively parallel sequencing which have a very high negative predictive value and have significantly reduced the need for invasive procedures, though invasive tests are still required to confirm a positive result (110-113). The wider clinical application of these techniques and their possible incorporation into national programmes are under consideration (114).
The fetus with sonographic abnormalities and a normal karyotype remains a diagnostic challenge. Recently, array CGH has been introduced to prenatal diagnosis based on its superior sensitivity for chromosomal imbalances (115-118). One of the main challenges of the use of array CGH in this setting is the potential difficulty in interpreting the significance of some CNVs (119, 120). It is particularly important in prenatal cases to discern between incidental findings and chromosomal variants that are relevant to the fetal abnormalities. In that context, cytogenetic laboratories have assumed a stepwise approach to evaluate these findings (121). Parental tests are used to establish whether the CNV has been inherited as its identification in an unaffected parent makes it less likely to be relevant. Moreover, the region of imbalance is assessed for the presence of relevant genes and databases, such as the Database of Genomic Variants, are assessed to determine whether it has been previously reported in association with a syndrome or specific phenotype or whether it has been reported as polymorphic.
More on the topic Genetic aspects of the clinical encounter:
- Arulkumaran S., Ledger W., Denny L., Doumouchtsis S. (eds.). Oxford Textbook of Obstetrics and Gynaecology. Oxford University Press,2020. — 928 p., 2020
- Chapter 50 Ovarian and Adnexal Disease