Basic genetic concepts in clinical practice
Our understanding of how genetic information is organized within cells, the way it underlies development and function by directing protein synthesis, and how it is transmitted from generation to generation has relied on our deciphering of chromosomes and genes.
Within the field of human genetics, this broadly translates into the cytogenetic (chromosomes) and molecular genetic (genes) approaches. The DNA double helix is wound around histone molecules constituting the chromosomes that are visible on light microscopy under certain conditions whereas the study of the nucleotide sequence within genes themselves is beyond the resolution of the microscope and requires the use of specific molecular techniques. In some ways, the difference between cytogenetics and molecular genetics is one of scale: a deletion of a small chromosomal segment may lead to the loss of multiple genes whereas a point mutation only affects one gene. Recently, however, the distinction between these two disciplines has been increasingly blurred by the development of newer techniques and the emergence of the so-called molecular cytogenetics that has greatly augmented the resolution of traditional cytogenetic approaches (1, 2). Moreover, genetics has been revolutionized by the emergence of novel approaches such as next-generation sequencing and the evolution of these techniques has transformed the impact of genetics on clinical practice (3-7).Genetic variation
The application of genetics in clinical medicine is largely based on the study of genetic variation. It is very important that terminology is used accurately and in the right context otherwise it can lead to confusion and misinterpretation. Changes in the sequence of bases within a gene do not necessarily cause problems and the term variant of unknown significance denotes genetic variation with an undetermined impact on gene function.
The term polymorphism refers to genetic variants that are relatively common within specific populations, have no known detrimental impact, and are considered to be variations of normality. On the other hand, the term mutation or pathogenic mutation describes genetic variants that affect gene function and alter the phenotype. It is important to use these terms accurately and consistently and interpret them correctly when they appear in the reports of genetic tests.When referring to individual genes, variants are categorized as substitutions, deletions, or insertions and can occur either within the coding sequences of genes or the non-coding regions (introns or regulatory elements). Though most clinically important mutations are located in the part of the genome that is translated into proteins, non-coding variants can affect gene function by disturbing transcription and post-transcriptional modification (8-10). Within the coding region, a single base substitution may not alter the corresponding amino acid based on the degeneracy of the genetic code (synonymous). On the other hand, substitutions may lead to the incorporation of a different amino acid in the polypeptide chain (missense) or the introduction of a stop signal in which case protein synthesis is terminated altogether (nonsense).
Variants of unknown significance within coding sequences are most likely to be of the missense variety. The impact of substituting one amino acid for another within a polypeptide chain will be determined by a number of factors including the physicochemical similarity between the two amino acids, the location of the substitution within the chain, and the functional role of that part of the protein and the segregation of the variant with the relevant phenotype in the family b eing investigated (11-14).
Deletions or insertions involve one or more nucleotides, exons, or the entire gene. When chromosomal regions are involved, many genes may be affected. Referring to smaller-s cale variants, these can be described as either in-frame or out-of-frame depending on whether the number of deleted/inserted nucleotides is a multiple of three.
If it is, the overall reading frame of the gene is maintained (as the code is read in triplets) and a protein is synthesized even though there will be loss or introduction of amino acids in the polypeptide chain. If it is not a multiple of three, the reading frame shifts and the process is deranged.Epigenetic regulation
Epigenetics deals with influencing gene function without altering the gene's sequence of bases. Epigenetic mechanisms underlie the differential expression of genes in different tissues and, in doing so, underpin development and tissue specification (15). The pattern of selective silencing of certain genes and transcriptional activation of others is mediated by specific protein complexes and is stably inherited through mitotic divisions unless specifically erased and reprogrammed. Gametogenesis and early embryo development are particularly important periods in epigenetic programming. The main epigenetic mechanisms are DNA methylation which involves the transfer of a methyl group to the cytosine of a CpG dinucleotide, modification of histone molecules which results in genes becoming more or less accessible for transcription, and non-coding RNAs which are RNA molecules that do not get translated and play key regulatory roles at multiple steps including transcription, post-transcriptional modification, splicing, and translation (16-19).
Genomic imprinting is a well-described example of epigenetic modification and is the process, established during gametogenesis, which ensures monoallelic expression of specific genes in the zygote based on differential gene silencing according to the parent of origin (20-22). There is strong evidence that imprinted genes play an important role in the regulation of fetal growth, shedding new light on this key aspect of pregnancy (23, 24). Techniques of assisted reproduction have been associated with an increased risk of imprinting disorders such as Beckwith-Wiedemann syndrome and this has been attributed to a possible disturbance of the imprinting process (25-27).
However, it is not clear whether this is a result of the technique itself or the underlying aetiology of infertility (28, 29).Applied cytogenetics
The technique of karyotyping, which allows the visualization of chromosomes of cultured cells that are arrested in metaphase, has been the mainstay of cytogenetic investigations. Chromosomes are stained to produce a characteristic banding pattern with Giemsa stain being widely used (G-banding) (30, 31). Apart from diagnosing numerical chromosomal abnormalities such as aneuploidy and trip- loidy, the technique can identify structural rearrangements such as translocations and ring chromosomes as well as segmental deletions, duplications, or inversions, and can pinpoint their position on specific chromosomes based on the bands involved (Figure 4.1). Its usefulness is limited by its sensitivity which depends on the size of the chromosomal segment involved and the skill of the operator. Though the technique has evolved and its resolution has improved over the years, it can still only detect abnormalities which are about 5 million base pairs (Mb) in length or greater.
The technique is particularly useful in identifying balanced chromosomal rearrangements such as translocations or inversions even if these rearrangements do not result in the net loss or gain of genetic material, an advantage over newer molecular techniques that rely on quantification rather than visualization. In the context of obstetric practice, the karyotype remains the basis of genetic assessment of a fetus following interventional prenatal techniques such as chorionic villous sampling and amniocentesis. It is also the technique of choice in investigating couples presenting with recurrent early pregnancy losses or who have had a baby diagnosed with a chromosomal abnormality based on its ability to detect balanced rearrangements that can themselves predispose to unbalanced chromosome complements in the offspring.
Smaller, submicroscopic deletions (microdeletions) cannot be identified using the routine karyotype.
The technique of fluorescence in situ hybridization (FISH) uses fluorescently labelled DNA probes corresponding to specific DNA sequences which hybridize with the specific chromosomal segment and allow confirmation of the presence of this segment under the microscope (32) (Figure 4.2). In contrast to the karyotype, the clinician needs to ask the laboratory to carry out a targeted test using a specific probe, the implication being that this requires prior clinical suspicion. An example of this approach in prenatal diagnosis is the use of FISH to exclude 22q11.2 microdeletion syndrome in fetuses diagnosed with cardiac malformations (33). With the exception of chromosomal aneuploidies, this syndrome is one of the commonest causes of heart defects detected antenatally (34).The need to identify even smaller chromosomal deletions/dupli- cations was addressed by the development of a molecular cytogenetic technique known as array comparative genomic hybridization (array CGH) (35-37). It is based on differentially fluorescently labelling DNA from the individual to be tested and a reference DNA and then allowing both to competitively hybridize with an array of DNA segments representing the entire genome. Any missing or extra genetic material in the test DNA will disturb the comparative hybridization at the corresponding unbalanced regions resulting in a different fluorescent signal compared to the rest of the genome where test and reference DNA have no quantitative difference. The signal patterns are analysed by a computer which collates a genome-wide
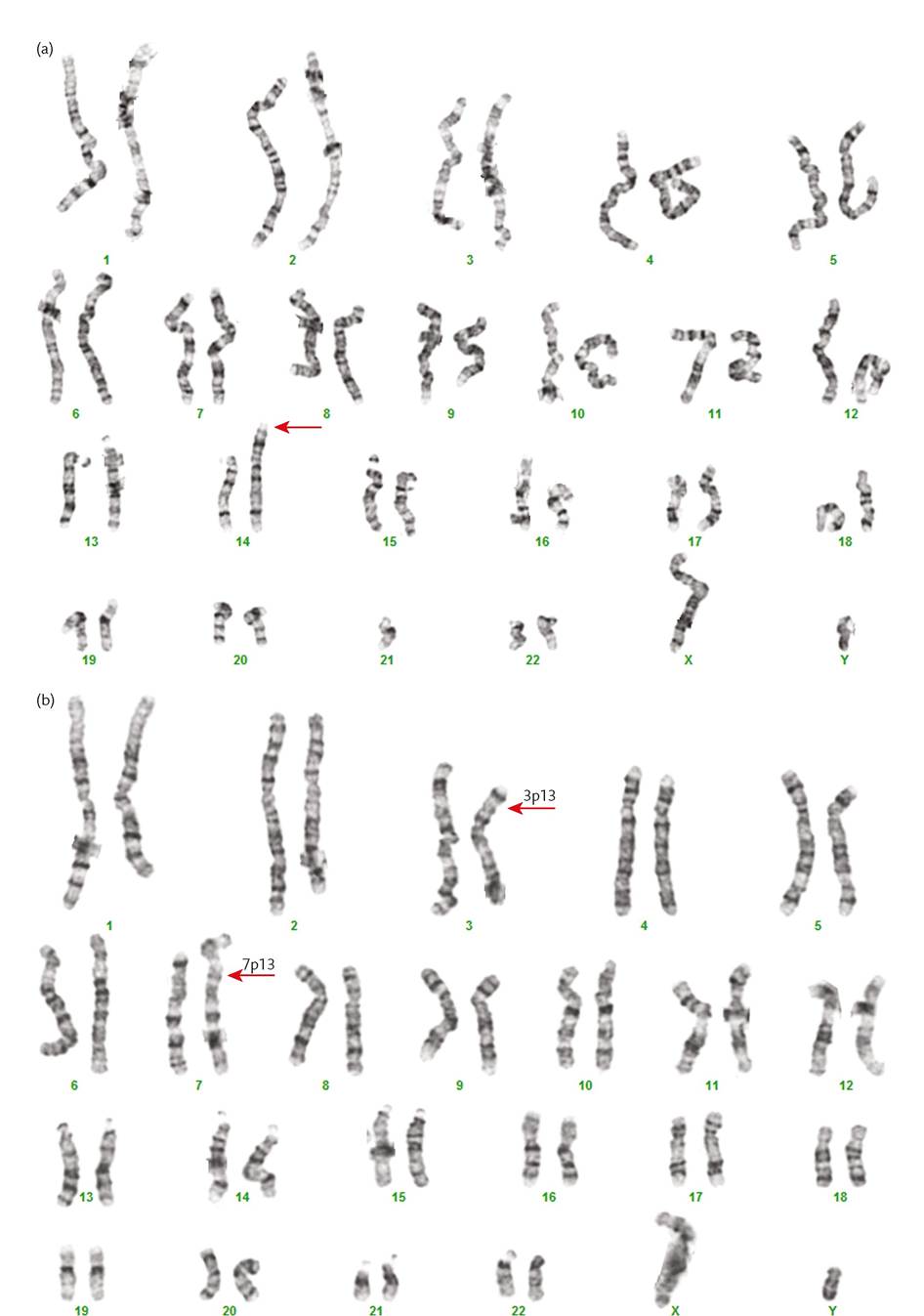
Figure 4.1 G-banded karyotyping showing a 14;21 Robertsonian translocation in (a) and a reciprocal translocation between chromosomes 3 and 7 in (b). The karyotype in (a) has 45 chromosomes as a result of fusion of the long arms of the acrocentric chromosomes 14 and 21 (arrow shows the derivative chromosome). In (b), there is an exchange between the short arms of one chromosome 3 and one chromosome 7.
The arrows denote the derivative chromosomes and the break points at bands 3p13 and 7p13. Both chromosome complements can give rise to genetically unbalanced gametes.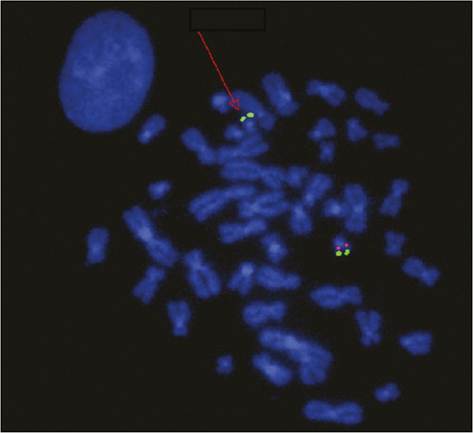
Figure 4.2 Fluorescent in situ hybridization showing a heterozygous 22q11.2 deletion. The chromosome with the deletion (arrow) exhibits the green signal for the chromosome 22 marker but lacks the red signal for the 22q11.2 locus which is seen on the homologous chromosome. The signals appear paired because chromosomes are in the form of duplicated chromatids at this stage of mitosis.
Courtesy Dr Carolina Sismani, Cyprus Institute of Neurology & Genetics.
The technique assesses the entire genome for imbalances and in many centres it has replaced the G-banded karyotype as the first- line investigation for patients with learning disabilities, dysmorphic features, and multiple congenital malformations (38-40). An inherent consequence of the technique’s high sensitivity is the need to assess the significance of small imbalances in the context of the specific clinical situation. Assessing the potential pathogenicity of such findings follows a number of steps including parental testing to determine whether this is a de novo or inherited variant. Array CGH does not detect balanced rearrangements and may not detect low-level mosaicism.
Applied molecular genetics
The basis of molecular genetic techniques is their ability to identify the sequence of nucleotides at a specific gene locus and, by comparing this with the normal reference sequence, identify genetic variants which may or may not be relevant to the clinical context in which tests are initiated. Frequently requested molecular genetic testing falls under two main categories: tests that seek to determine whether variation exists within a specific gene, and in doing so aim to establish a genetic diagnosis, and tests that are more limited in
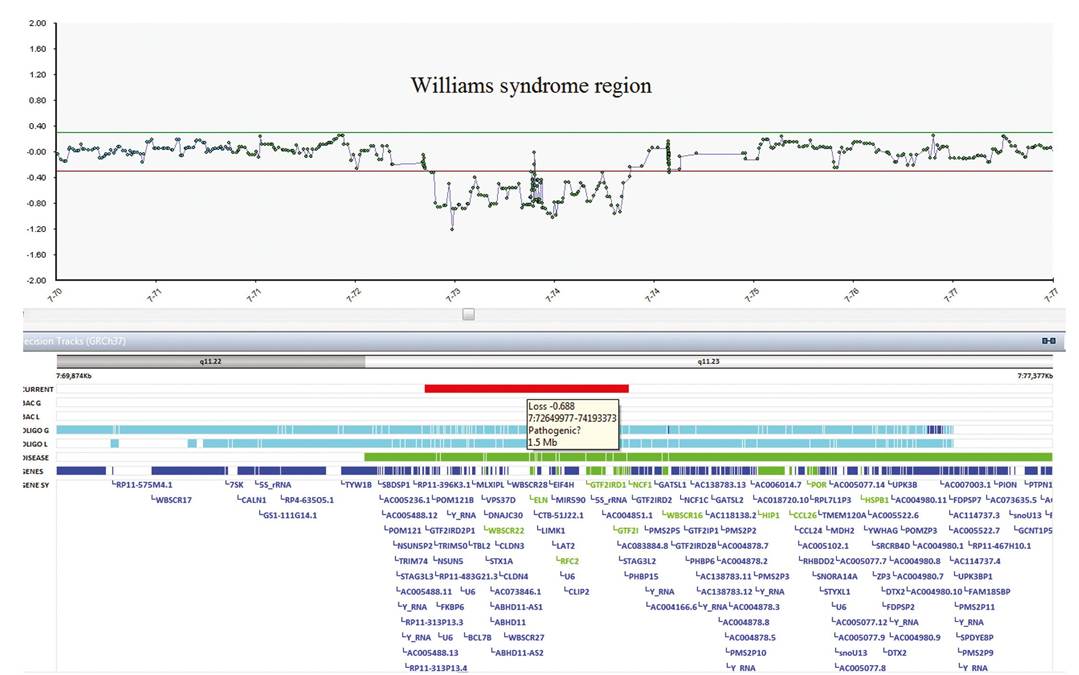
Figure 4.3 Array CGH result showing ratio of intensity of test and reference DNA in logarithmic scale (y axis) in relation to the genomic coordinates (x axis). In the logarithmic scale, deletions are represented by negative ratios. This result shows a deletion of approximately 1.5 Mb in length (thick red line) in the long arm of chromosome 7 which corresponds to the Williams syndrome critical region.
Gene sequencing is the centrepiece of modern molecular genetics with Sanger sequencing being considered as the gold standard (41). The technique uses DNA replication to determine the sequence of nucleotides in a single-stranded fragment of DNA. It relies on the introduction in the nucleotide mix (used in DNA replication) of additional modified nucleotides (containing the four bases) which lead to the termination of replication once they are introduced into the new chain (42). A computerized system determines the sequence of DNA bases by interpreting the position of this termination (Figure 4.4).
Sequencing techniques are not always best placed to detect specific types of mutations and partial or whole gene deletions/dupli- cations require a different approach. A polymerase chain reaction (PCR)-based technique known as multiplex ligation-dependent probe amplification uses the concurrent amplification of different gene segments (43). The resulting amplified segments have, by
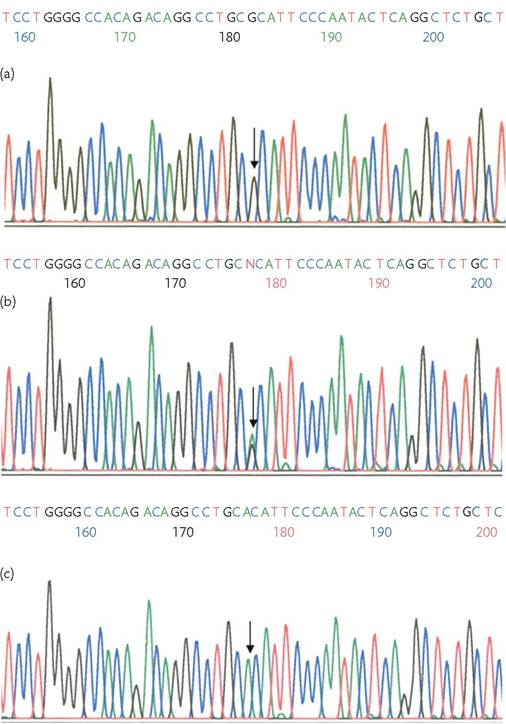
Figure 4.4 Sanger sequencing chromatogram. The peaks represent the four bases, each labelled with a different fluorescent dye, with the control sequence represented in (a). Guanine is substituted by adenine in a heterozygous state in (b), where both peaks are seen, and in a homozygous state in (c). The software sequencing numbers shown in this chromatogram do not correspond to base positions and differ between alignments.
Courtesy Dr Maria Loizidou, Cyprus Institute of Neurology & Genetics.
CHAPTER 4 Genetics for the obstetrician and gynaecologist design, different sizes and can be separated by electrophoresis and visualized based on their fluorescent labels. The various signal peaks produced correspond to the different gene segments and a comparison with a reference sequence allows identification of parts of the gene that are either deleted, resulting in a reduced signal, or duplicated, resulting in an increased signal (Figure 4.5).
Sequencing techniques are also widely used to determine the presence of a specific mutation though this can also be achieved by techniques targeting the specific position within the gene. In PCR- based techniques, amplification is dependent on the presence of the mutation and can be detected using fluorescent labelling (44, 45).
While the techniques previously described continue to constitute the basis of molecular genetic investigations in many settings, the emergence of next-generation sequencing has revolutionized genetic testing (46-48). It relies on a technique known as massively parallel sequencing which is based on genome fragmentation and parallel sequencing of fragments using novel sequencing approaches that allow rapid identification of incorporated bases during replication of DNA templates. The sequence data is then contrasted against a library of reference sequences and discrepancies are noted. The efficiency of this technology has made the assessment of the entire exome and genome available for the investigation of disease and has led to the identification of the genetic basis of many monogenic disorders that had eluded molecular characterization (49, 50).
More on the topic Basic genetic concepts in clinical practice:
- Introduction
- Arulkumaran S., Ledger W., Denny L., Doumouchtsis S. (eds.). Oxford Textbook of Obstetrics and Gynaecology. Oxford University Press,2020. — 928 p., 2020
- References
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025
- Antiretroviral Therapy Today
- Chapter 44 Cell Biology and Principles of Cancer Therapy