DEVELOPMENTAL LIMB DISORDERS
Maximum effect of in utero fetal position and pressure is obvious on limbs, leading to torsion and angular alignments of long bones as well as some extent of joint and muscle contractures.
Even normal newborns have 20-30 flexion contractures at hip and knee, which disappears by 4-6 months of age. These deformities tend to be exaggerated in primi mothers and oligohydramnios.Primary challenge in developmental limb disorders is to differentiate between physiological and pathological aberrations for rational management
Congenital limb malformations are relatively uncommon and include:
Limb deficiency amelia (complete absence of limbs), Phocomelia (absence of proximal and middle segments, with hands/feet directly attached to trunk), Hemimelia (absence of distal and middle segments, with preserved arms/thighs), Acheiria (absence of hands/feet) and Adactylia (absence of fingers or toes).
While fetal thalidomide exposure was once the commonest cause of limb-deficiency defects, currently most of these malformations are sporadic and isolated defects, probably representing intrauterine amputation of developing fetal limb by amniotic constriction bands.
Partial or minor limb defects are also seen in rare sporadic syndromes, e.g. Holt-Oram syndrome (with CHDs), Cornelia de Lange syndrome, etc. Radial club hand (complete or partial absence of radius) is a rare but important limb defect, seen in VATER association (Chapter 12.19) or Holt-Oram syndrome.
Amniotic constriction bands (annular bands or constriction rings) due to entrapment of affected part in amniotic membranes are common over toes and fingers, but may also involve whole or part of limb circumference. Severe constriction rings may cause auto-amputation of distal parts (Fig. 25.2).
Digit abnormalities, e.g. polydactyly (super-numerary digits) or syndactyly (fusion of digits) are common and usually cosmetic birth defects.
However, some of them are associated with other malformations, e.g. craniosynostosis, chromosomal trisomies, fetal- hydantoin syndrome, cornelia de lange syndrome or oro-facial digital syndrome. Surgical excision of well- formed extra digit is preferred after infancy, though rudimentary digit may be ligated at birth to allow autoamputation (Fig. 23.2).Brachydactyly (short, stubby fingers/toes) or Clino- dactyly (incurving of usually little finger) are minor phalangeal abnormalities, common in chromosomal disorders, e.g. Down syndrome, Rubinstein-Taybi syndrome, Turner syndrome, etc.
Congenital hemihypertrophy, i.e. unilateral overgrowth of whole or part of the body, may be associated with mental retardation, Wilms' tumor or adrenal
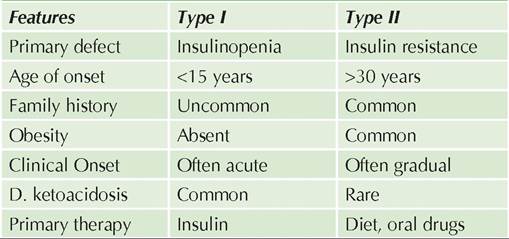
Fig. 23.2: Polysyndactyly.
carcinoma. Underlying tissues are normal in congenital hemihypertrophy, unlike acquired hemihypertrophy in neurofibromatosis, hemangioma or lymphangioma.
Fixed flexion deformities, limited to hand/feet are not uncommon due to stenosed tendon sheath and usually present with Trigger thumbs/fingers at terminal interphalangeal joint of hands and Hammer toe at proximal interphalangeal joint of toes. Although passive stretching/strapping may be useful in young infants, surgical release is often necessary to achieve full function.
Arthrogryposis multiplex congenita is a symptom complex characterized by multiple joint contractures at birth, leading to severe limb deformities. Though seen in many syndromes, commonest prototype is Amyoplasia, accounting for ~40% cases and discussed here.
Etiology is exactly unknown, suspected to be due to a fetal neurological defect with decreased number of anterior horn cells or fetal myopathy, leading to decreased fetal movements and subsequent in utero contractures.
Clinically, joint contractures are present in all limbs, more prominent in lower limbs, though variants with are known.
Although contractures are essentially nonprogressive, many cases die in infancy due to respiratory muscle involvement.Diagnosis is clinical, followed by radiological evaluation for major orthopedic defects, e.g. spinal deformities and hip dislocation.
Treatment aims to achieve maximum function and reduce cosmetic deformities, with physiotherapy, orthotic support and surgical correction of major defects.
Incidentally, a mythological character in Mahabharat 'Astavakra' was probably a case of Arthrogryposis multiplex congenita.
Developmental dysplasia of hip (DDH), also referred as congenital dislocation of hip (CDH), is characterized by partial/complete and temporary/permanent dislocation of femoral head from its acetabular cavity, affecting stability of hip joint.
Etiology: Stability of hip joint depends on depth and configuration/angulation of acetabulum and strength of surrounding ligaments/muscles.
DDH may result from: (a) underdeveloped shallow acetabulum or small/anteverted femoral head, (b) ligamental laxity, due to neuromuscular disorders or maternal estrogen effect, or (c) abnormal hip posture during/after delivery, which dislocates the hip and keeps it dislocated.
Early DDH is usually relocatable, but subsequent contractures in long-standing cases turn it into a permanent deformity.
Epidemiology: DDH is more common in females (9:1), first-born child (~60%), full term breech deliveries (30-50%) and in presence of similar family history (? hereditary). It is relatively less common in India than in western countries, probably due to the Indian practice of carrying the child on waist with abducted hips that prevents dislocation of hip despite anatomical abnormality.
Clinically, DDH may be bilateral or unilateral, usually on left side (60%) or hip is usually normal at birth and dislocates postnatally on flexion/adduction of hip, with following signs:
• Asymmetrical skin folds in groin/thigh;
• Restricted abduction at hip, leading to difficulty in changing of diapers; (Fig.
23.3A)• Galeazzi sign, i.e. apparent shortening of limbs with uneven knee-level, when knees and hips are flexed in supine position;
• Barlow test is conducted to assess stability of undisplaced hip, by placing the baby in supine position and stabilizing the hips in flexion and adduction. When a posterior force is applied by keeping thumbs medially and fingers externally, an audible click or palpable clunk is felt as femoral head moves out of acetabular cavity. Similar click/clunk also appears after removal of force, due to relocation (Fig. 23.3B).
• Ortolani test is used to decide whether CDH is relocatable. When hip is flexed and gradually abducted in supine position, femoral head is lifted anteriorly due to lever action and may enter back into acetabulum with a clunk (no click), indicating relocatable hip. Ortolani test is often negative after two months as hips become un-relocatable due to secondary contractures (Fig. 23.3C).
• Functional defects, e.g. limping or waddling gait are late features of DDH.
International Hip Dysplasia Registry (IHDR) has recommended following nomenclature for severity spectrum of DDH—(a) dysplasia, (b) subluxable, (c) dislocatable, (d) dislocatable but reducible, and (e) dislocatable and irreducible.
Diagnosis is confirmed on pelvic X-rays in normal and frog-position, though ultrasonography is preferred choice in first six months of life till femoral head is ossified, suggestive of acetabular dysplasia, hip dislocation and abnormal femoral head anatomy. CT/MRI
Fig. 23.3: Clinical assessment for DDH in newborns: (A) Limited abduction on affected side; (B) Barlow (dislocation) test; (C) Ortolani (relocation) test.

Fig. 23.4: X-ray: Developmental dysplasia of hip.
Note flat acetabulum on right side with increased joint space and Upward-outward dislocation of femoral head.
(Courtesy: Dr S Shrivastava)
may be used in difficult cases. Many cases may not be obvious at birth and need repeated evaluation.
X-ray pelvis (AP view) indicates possibility of DDH, if (Fig 23.4):
• Femoral head is above the Hilgenreiner's line, a horizontal line drawn through right and left triradiate cartilage,
• Femoral head is medial to the Perkin's line, a perpendicular line to Hilgenreiner's line through a focus at the lateral side of the acetabulum.
• Disruption in Shenton's line, a smooth arc that connects the femoral neck to the superior margin of the obturator foramen.
• Acetabular index gt;35% at birth, defined as intersection between Hilgenreiner's line and a line drawn tangential to the lateral ossific margin of the roof of the acetabulum.
Management depends on age of diagnosis and whether hip is still relocatable.
• Relocatable hip: When diagnosed at birth or in early infancy, hip is often relocatable. These cases require manual relocation followed by splinting (Von-Rosen splint or Pavlik harness) in same position, i.e. flexed and abducted position for 1-2 months, which helps in acetabular development by femoral pressure, normalization of anatomical relationships and strengthening of surrounding ligaments to prevent future dislocations.
• Non-relocatable hip, usually diagnosed at 6-18 months may require:
- Splinting of hips at gt;90° flexion and 50-70° abduction, which gradually relocates femoral head into acetabulum within 3-4 weeks. Splinting is highly effective (~90%) in young infants and should continue till normalization of radiological relationship.
- Closed reduction after prior traction over femoral head to relocate it in acetabulum, followed by splinting may be attempted in older children, if simple splinting fails.
- Surgical open reduction is often required in older children gt;18 months and involves pelvic/femoral osteotomies and realignment.
Outcome depends on the age of diagnosis and efficacy of treatment.
Apart from gait or locomotion abnormalities, common complications of untreated CDH include avascular necrosis of femoral head and residual acetabular dysplasia.Limb-Angulation Disorders
In utero, a fetus rests in a position of complete flexion, with external rotation of femur and internal rotation of leg bones (bowing effect). Consequently, physiological bowing (Genu varum) is normal till 18 months of age. Later as the child starts to walk, weight bearing may lead to overcorrection of this angulation with development of physiological knock knee, which may persist till 5-6 years.
Genu valgum (Knock-knee), i.e. inward angulation of knee due to outward deviation of longitudinal axis of tibia/ femur, presents with abnormally divergent legs/ankle and awkward gait, while walking (Fig. 23.5). Severity of knock-knee is assessed by inter-maleollar distance at
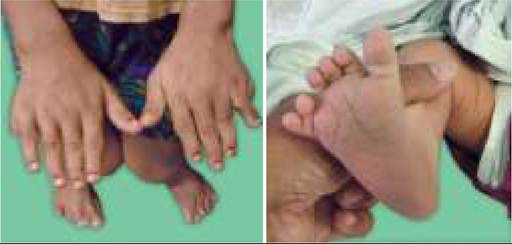
Fig. 23.5A and B: Limb angulation disorders: (A) Bow legs; (B) Knock knee.
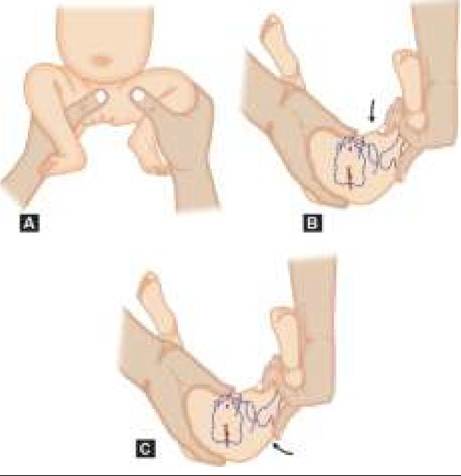
Fig. 23.6: Congenital talipes equinovarus (CTEV).
ankle, in straight supine position after approximation of knees, normal being lt; 8 mm.
Genu valgum is physiological from 18 months to 5 years of age. Severe or pathological genu valgum may be caused by: (a) rickets, (b) neuromuscular disorders, e.g. poliomyelitis or cerebral palsy, and (c) post infective/ traumatic damage to lateral knee cartilages. Most cases improve spontaneously with further growth and surgical correction is indicated only in progressive defects with functional impairment.
Genu varum (Bow-legs) is the reverse of knock-knee, with outward angulation of knee joints when ankles are approximated (Fig. 23.5).
Physiological bow-legs are common in infancy, but severe or pathological bow-legs may be due to: (a) rickets, (b) postural abnormalities (? excess use of round walkers) and (c) post-infective/traumatic damage to medial knee cartilages.
Severity of bow-legs may be assessed by measuring inter-knee distance with approximated ankles. Orthotic or surgical correction with osteotomies is required only in very severe cases.
Idiopathic tibia vara (Blount disease) is an important cause of progressive genu varum, specially in obese children, due to idiopathic and abnormal growth of medial aspect of proximal tibial epiphysis.
Genu recurvatum, i.e. hyp erextension of knee with a tendency to backward dislocation may be due to: (a) developmental ligamental laxity, (b) neuromuscular disorders (polio, cerebral palsy), or (c) post-traumatic/ infective injury to tibial tubercle or anterior knee epiphyses. Severe defects with recurrent dislocations may need splinting of knee or operative reduction.
Congenital Talipes Equinovarus (CTEV) or clubfoot is a common foot deformity (1:1000 births), characterized by plantar flexion of talus, inversion of calcaneus and adduction of forefoot.
Etiologically, CTEV may be (a) congenital or familial,
(b) postural, i.e. abnormal fetal-leg position with increased uterine pressure, e.g. oligohydramnios, or
(c) teratologic, e.g. in fetal neuromuscular disorders.
Clinically, it is more common in males (2:1) and with positive family history (? Hereditary). Most cases (2/3rd) are unilateral, more common on right side and present at birth with plantar flexion at ankle (varus) and adduction with internal rotation of foot (equinus), with soles facing inward (Fig. 23.6). Missed or untreated cases in early infancy may develop delayed walking, unsteady gait, callosities and disuse calf atrophy.
Diagnosis is clinical, though radiological evaluation in weight-bearing position is needed before orthopaedic correction, to assess extent of misalignment. CTEV may be detected on prenatal USG by 14-16 weeks.
Management, often termed as Ponseti method, includes:
• Conservative treatment should begin at birth in all cases, with instructing the mother to manipulate the deformity, as frequently as possible. Most cases require: (i) repeated manual correction, followed by taping/malleable splinting, or (ii) serial plastercast application in corrected posture (revised every 1-2 weeks). After correction, foot may be kept in Dennis-Brown splint till walking begins, to prevent re-misalignment.
• Surgical treatment is indicated in cases without clinical/radiological correction beyond 3 months of age, involving achilles tenotomy followed by placing a final cast for 3 weeks. After removal of the cast, foot has to be kept in a brace or Dennis-Brown splint-a foot abduction orthosis with two shoes or splints connected by a bar to holds the feet shoulder width apart. This brace needs to worn throughout the day for 3 months followed by only at night time until the child is 4-5 years old. Recurrence is high (80%) in non-compliant cases during brace phase, while it is only 6% incompliant cases.
Talipes calcaneovalgus is milder and opposite variant of club-foot, characterized by dorsi-flexed ankle with abducted and everted forefoot. Most cases are due to abnormal intrauterine position and correct spontaneously by 6-12 months.
Congenital metatarsus adductus or varus (adducted and supinated foot) is more common in first-born infants and young children, due to higher intra-uterine pressure in primigravida. Clinically, foot is convex laterally with prominent base of 5th metatarsal. In long-term, this defect may cause in-toe gait and abnormal shoe wear. While passive exercises and corrective shoe-wear are enough in most cases, rigid deformities may require serial plastercasts or surgical correction.
Adolescent bunions (Hallux valgus deformity), although manifest in adolescence, is usually a result of congenital malalignment of first metatarsal with enlarged medial aspect of its head. It is more common in females and those with similar family history. Use of appropriately shaped shoes with wide toe-box is often enough, though orthotic correction or surgery may be needed in selected cases.
Rocker-bottom foot (congenital vertical talus) is an uncommon variant of club foot, due to abnormal axis of talus, characterized by hind-foot equinovalgus, convex planter surface and fore-foot abduction with dorsiflexion. It is usually associated with trisomy 13 (Edward syndrome) or arthrogryposis multiplex congenita.
Foot-arch abnormalities: At birth, foot-sole is normally flat due to lax ligaments and medial pad of fat. As the child starts walking, transverse and longitudinal arches develop for better stability, maintained by tarsal and metatarsal bones, planter ligaments and posterior tibial muscles. Most important is the medial longitudinal arch that is well formed by 3 years and tends to shift body weight laterally.
Pes planus (Flat foot) denotes absent or poorly formed medial longitudinal arch, leading to abnormal weight bearing on medial rather than the lateral arch.
Etiologically, while physiological till 3 years, pathological flat-foot may be due to—(a) ligamental laxity/ injury (flexible flat-foot), or (b) tarsal coalition, i.e. congenital or post-infective/traumatic fusion of tarsal bones (rigid-foot).
Clinically, flexible flatfoot presents in toddlers with unsteady gait with heel in vagus and forefoot in an abducted position but arch is normal in non-weightbearing position. On the other hand, rigid-foot usually manifests in adolescents with foot/peroneal pain during walking and foot remains flat in weight-bearing as well as non weight-bearing position. Shoe wears out faster on the medial side of the sole.
Tip-toe walking helps to differentiate flexible flatfoot from rigid flatfoot. In a flexible flatfoot, even though the medial longitudinal arch is collapsed during the stance phase; the arch is formed and the heel swings into varus on tip-toe walking but not so in a rigid flatfoot.
Diagnosis is clinical with X-rays in weight-bearing position, which may also demonstrate tarsal coalition.
Treatment is unnecessary for flexible flat-feet except orthotic support and exercises in symptomatic cases. Rigid-foot may require orthotic support or even surgical correction.
Pes cavus, the reverse of pes planus with exaggerated medial longitudinal arch, is usually an acquired abnormality during mid-childhood, due to neuromuscular disorders, e.g. Friedreich's ataxia. Conservative treatment is not beneficial and surgical reconstruction of arch is usually necessary.
Developmental upper limb deformities are relatively uncommon than those of lower limbs, though important ones are as follows:
Sprengel deformity denotes developmental failure of the scapula to descend to its normal location. Clinically, the scapula is located at an abnormally high position (winged scapula), with/without associated features, e.g. webbed neck, low hairline and Klippel-Feil anomaly (Chapter 23.2.3). Severe cases may cause significant cosmetic defect or restricted shoulder mobility, which requires surgical correction.
Nursemaid elbow (Pulled Elbow) denotes sudden subluxation of radial head with partial entrapment of annular ligament in radiohumeral joint, usually seen in infants and toddlers due to relatively less bulbous radial head. Subluxation usually begins following jerky longitudinal traction over extended and pronated forearm during play or when the child suddenly falls while hand is held by parents.
Clinically, these cases present with sudden onset of crying and refusal to move upper limb, with hand held in prone position (psuedoparalysis). The arm is held flexed at the elbow with complains of pain at the elbow or the wrist. Baby will allow passive flexion and extension of the elbow but will resist rotations. There are no signs of inflammation around the elbow and X-ray is normal.
Management involves reposition of the radial head and annular ligament by either of the following two methods:
• Pronation Maneuver with—(a) holding the arm in semi-prone position with 90° flexion at elbow, (b) simultaneous pronation of forearm and extension of elbow, and (c) flexion of the elbow with forearm pronated; or
• Flexion-supination maneuver with—(a) holding the arm in full supination, (b) hyperflexing the elbow with pressure over radial head, to move it inside the annular ligament, when a click is felt over the lateral aspect of elbow.
Once reduced, the pain relief is dramatic with return of functions. No further immobilization is required.
Recurrent subluxation is common in 20-30% cases, till maturity of radial head by 3-4 years. Parents must be counseled not to grab the child by the arm or hand but rather pick them from axilla.
Growing Pains
Growing pains (Recurrent limb pains) are common between 5 and 12 years of age with estimated prevalence of 10-20%.
Etiology is not established but a behavioral component or transient post-activity muscle edema within the limited facial space has been suggested. Other possible reasons include low pain threshold, postural problems, hypermobility of joints, emotional issues or hypovitaminosis D.
Clinically, these cases present with recurrent pain in calves, shin and thigh area, usually in evenings or night. Pains are typically more common after strenuous daytime activity and not associated with local objective signs, e.g. swelling.
Diagnosis is clinical, which may be supported by Peterson criteria, i.e.: (a) intermittent pain once or twice a week, (b) asymptomatic phase between two episodes, (c) short-lasting and poorly-localized limb pains, and (d) excluding unilateral limb pain or the nocturnal pain that continues till next morning.
Presence of fever, weight loss, limp between the episodes or local tenderness indicates need for exclusion of other causes, e.g. benign joint hypermobility syndrome, juvenile idiopathic arthritis, complex regional pain syndrome as well as local osteo-articular trauma, infection or malignancy.
Management: No treatment is necessary except assurance, local rubbing/hot fomentation and mild analgesics, e.g. paracetamol. Child may benefit from simple stretching excises of quadriceps, hamstrings and gastrocnemius group of muscles. Gradually, the child outgrows these pains, with no long-term effects.
23.2.2