HYPERPIGMENTED LESIONS
Hyperpigmentation denotes increased melanin production and/ or deposition in epidermis (brownish black) or dermis (bluish-black), presenting either as isolated skin lesions or as a part of systemic disease (Table 25.3).
Common hyperpigmented lesions in children are as follows:Cafe-au-lait spots are dark-brown, well-demarcated, macular lesions of variable size (see Fig. 18.17), present at
TABLE 25.2: Common pigmentary lesions at birth
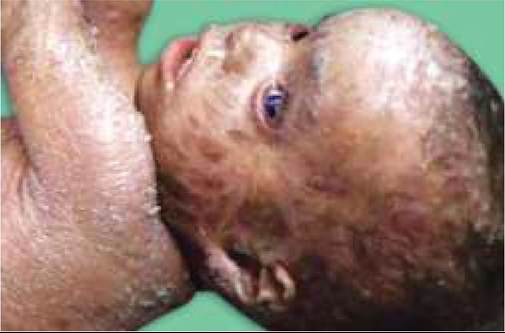
*or mastocytoma, which may blister
TABLE 25.3: Common hyperpigmented skin lesions
• Primary skin lesions:
- Epidermal: Cafe-au-lait spots, freckles, Ientigines
- Dermal: Mongolian spots
- Mixed: Post-inflammatory, incontinentia pigmenti
- Melanocytic nevi: Congenital or acquired
• Secondary to systemic disease:
- Addison disease
- Kala-azar
- Infantile tremor syndrome
birth or appear later on. Although these spots are present in ~10% of normal children, two important syndromes associated with many or large-sized cafe-au-lait spots are:
• Neurofibromatosis (Chapter 18.14), associated with gt; 5 spots of gt;5 mm; and
• McCune Albright syndrome with large asymmetrical cafe-au-lait spots of irregular borders, polyostotic fibrous dysplasia of bones, precocious puberty and multiple endocrinopathies.
Ephelides or freckles are brown macules of lt;3 mm, commonly seen in school children over sun-exposed parts, e.g. face, upper back and arms/hands. These lesions tend to appear in summer and fade in winter season. Except for a possible association with melanoma, these lesions are essentially benign.
Lentigines, though appearance-wise similar to freckles, these lesions may develop on any part of body, unrelated with sun exposure and remain permanent. Multiple lentigines may be indicative of Addison's disease or Peut- Jeghers syndrome (with GIT polyposis).
Mongolian spots are bluish/grayish flat patches (confused with bruises) of variable size mainly over sacral area and upper thighs (Fig. 12.6). These spots, seen in ~80% of newborns, represent presence of melanocytes in dermis, due to incomplete migration from neural crest to epidermis in embryonic life. Mongolian spots usually fade away in later life and are of no clinical consequence. Melanocytic nevi, an abnormal but benign clustering of normal melanocytes usually at epidermal-dermal junction, may be congenital in 1% newborns or acquired usually during or after adolescence. Congenital melanocytic nevi are usually larger than acquired lesions and carry a small risk of developing melanoma in later life. Malignant transformation is extremely rare in acquired nevi.
Congenital giant pigmented nevi (gt;20 cm), most commonly seen over back are of special significance due to frequent association with leptomeningeal melanosis and predisposition for subsequent malignant transformation. Clinically, these lesions are asymptomatic or present with neurological manifestations, e.g. hydrocephalus, mental retardation and/or seizures. MRI of brain is essential in all cases. Local excision and grafting reduces the risk of secondary melanoma but at the cost of disfigurement.
25.4.2 Hypopigmented lesions_______________
Primary hypopigmented lesions are caused by congenital or acquired deficiency in number of melanocytes and hereditary defects in tyrosine metabolism, though similar lesions are also seen in fungal infections (Table 25.4). Some common hypopigmentary disorders are as follows: Albinism, a congenital hypopigmentation disorder, denotes “complete or partial failure of melanin production in skin, hair and/or eyes, despite the presence of normal number, structure and distribution of melanocytesquot;.
Etiologically, albinism is caused by many hereditary enzyme defects at different stages of tyrosine metabolism, responsible for biosynthesis or distribution of melanin.
Clinically, Albinism may be generalized or localized and severity may vary according to the enzymatic defects, though common manifestations include: (a) generalized or partial hypopigmentation of skin, hair and/or eyes, since birth, (b) decreased visual acuity, photophobia and strabismus, with red-eye reflex due to translucent Iris (Fig. 25.7A). Skin cancer and complete blindness are late complications in most severe forms of albinism and albinism-associated syndromes. (Ch 11.6.1)Management is non-specific, including: (a) protection from sunlight to prevent skin cancers by protective clothing, sun screens, sun glasses and caps, (b) correction of refractive errors, and (c) genetic counseling.
Vitiligo is an acquired (d/d albinism) and usually patchy depigmentary disorder, due to reduced or absent melanocytes.
Etiopathogenesis is unclear, though most probably involves increased destruction of melanocytes due to autoimmune or other unknown factors. Family history is present in 30-40% cases and many cases are associated with systemic autoimmune disorders, e.g. Addison's disease, pernicious anemia, thyroid disease, etc.
Clinically, most pediatric cases present after first decade of life with well-demarcated milky-white or
TABLE 25.4: Common hypopigmented skin lesions
Generalized:
Congenital : Oculocutaneous albinism
Acquired : Universal vitiligo
Localized:
Congenital : Partial albinism and associated syndromes
Acquired : Vitiligo
Leprosy
Fungal skin infections
Pityriasis alba
Post-inflammatory lesions
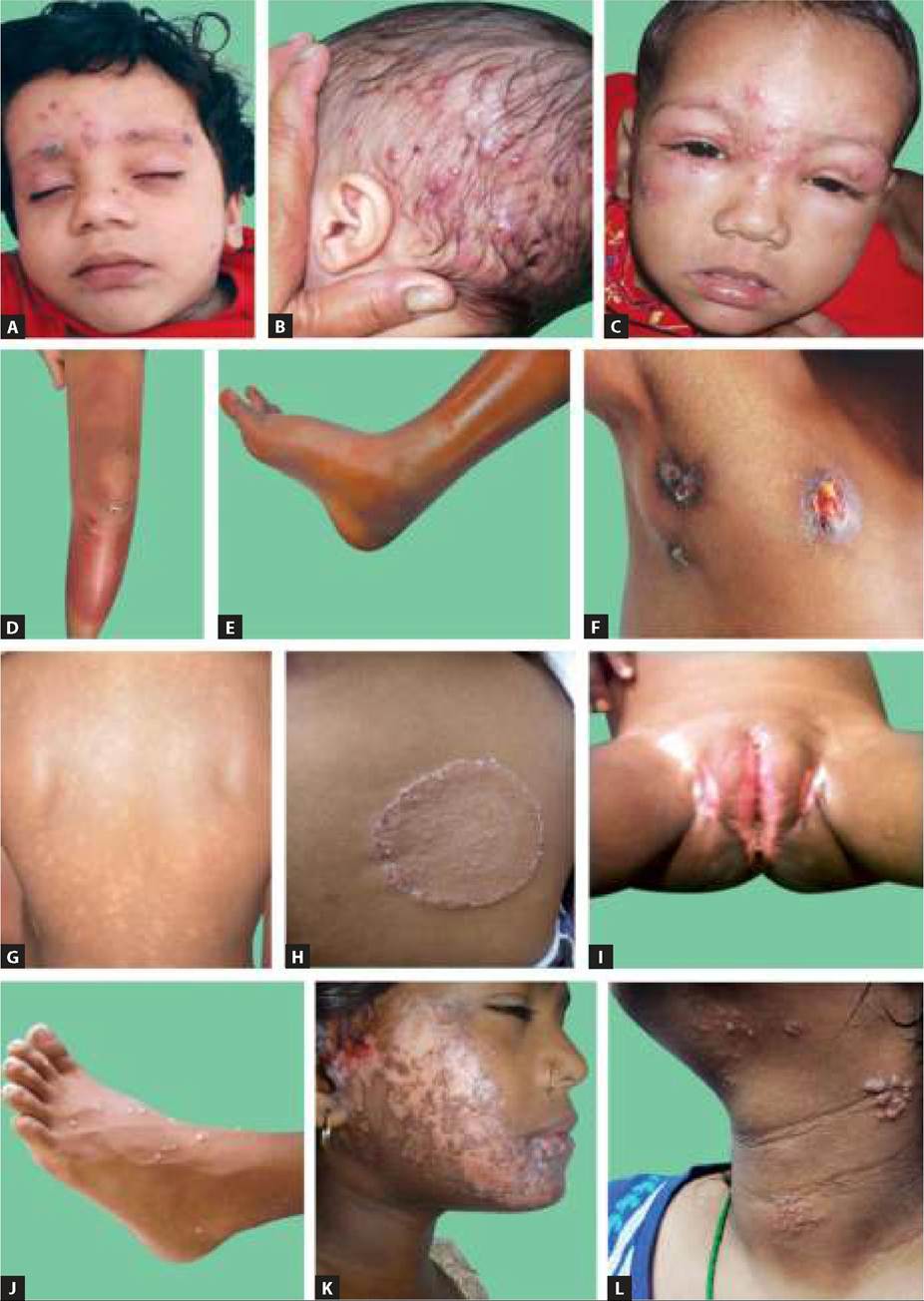
Fig. 25.7: (A) Albinism; (B) Vitiligo.
Hypopigmented patches, usually over normally hyperpigmented areas, e.g. face, axilla, groin and areola or friction-prone areas, e.g. distal extremities and joints (Fig. 25.7B). Vitiligo may be localized, segmental or generalized. Course of vitiligo is variable, with spontaneous re-pigmentation in 10-20% cases.
Diagnosis is clinical or on skin biopsy. Common D/D of vitiligo includes leprosy (sensory loss), albinism (congenital) and leukoderma (a non-progressive patch after burns or local inflammatory lesions.
Management: Reassurance regarding its benign nature is most important, considering social stigma attached to vitiligo. Management depends on the site and size of lesions and success rate is variable despite prolonged therapy.
Common modalities for small patches include: (a) topical steroids, e.g. fluocinolone acetonide or calcineurin inhibitors, e.g. Tacrolimus, (b) topical PUVA therapy in children above 10 years, and/or (c) cosmetic tattoo or skin grafting. Large lesions need systemic PUVA therapy. Systemic steroids are used to arrest progression of rapidly spreading lesions.
PUVA therapy consists of topical or systemic administration of psoralen (a plant-derived chemical, furocou- marins) followed by ultraviolet radiation to stimulate melanocyte function and regeneration. However, it is contraindicated in young children due to higher risk of skin cancers.
Pityriasis alba is the commonest hypopigmented skin lesion in children with unknown etiology, though often regarded as a mild form of eczema. Unlike general belief, it has no relation with calcium or iron deficiency.
Clinically, it is characterized by hypopigmented, round or oval, macular or slightly elevated patches with fine adherent scale and ill-defined margins over face, neck, upper chest and shoulders (Fig. 25.8). Itching is minimal or absent. Lesions are exacerbated by dryness and often
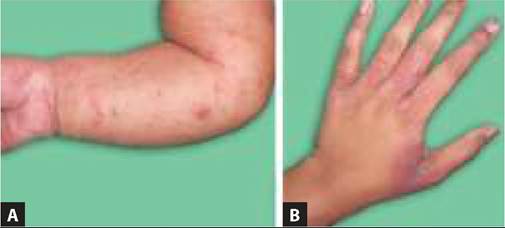
Fig. 25.8: Pityriasis alba.
run a waxing-waning course for many months before disappearing completely.
Diagnosis is clinical, though needs to be differentiated from T. versicolor or T. corporis by KOH preparation of skin scrapping.
Management: These are very benign and self-limiting lesions except cosmetic apprehension. Application of a lubricant or topical steroid (hydrocortisone 1%) thrice a day is often enough to ameliorate the lesion, though normal pigmentation may return after many months.
25.4