MUCOPOLYSACCHARIDES
Mucopolysaccharides or glycosaminoglycans (GAGs) are major constituents of connective tissue, including chondroitin sulfates, dermatan sulfate, heparan sulfate, keratan sulfate, hyaluronic acid and heparin.
Mucopolysaccharoidoses (MPS) are inherited defects in degradation of these GAGs, due to deficiency of specific lysosomal enzymes, leading to excess accumulation and consequent urinary excretion of various GAGs. Over 10 variants of MPS are known, each with a specific lysosomal enzyme defect, clinical presentation and severity (Table
11. 17). All of them are autosomal recessive, except Hunter syndrome, which is X-linked recessive. Some important types of MPS are discussed here:
Hurler syndrome (MPS IH) is the most severe and prototype manifestation of MPS with multiple organ involvement.
Clinically, These babies are normal at birth and usually manifest at 6 months-2 years of age with:
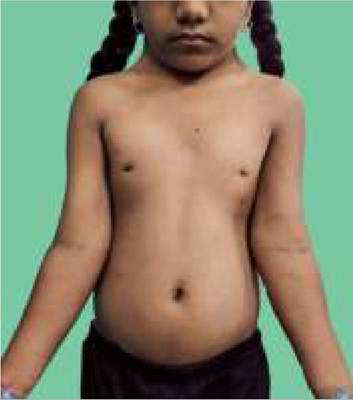
• Short stature and coarse facial features with prominent forehead, cloudy cornea, flat-broad nose, thick lips and large tongue (Fig. 11.10);
• Developmental delay with poor language development and hearing loss;
• Hepatosplenomegaly with protuberant abdomen and frequent umbilical/inguinal hernias;
Multiple bony deformities (dysostosis multiplex) include large, thick calvarium, dolichocephalic skull with shallow orbits, enlarged and deformed sella turcica, broad-spatulate 'oar shaped' ribs, anterior hypoplasia (beaking) of lumbar vertebrae with kyphosis, short and wide bullet shaped phalanges and deformed pelvis with short femoral head and coxa valga. Long bones have wide diaphyses, irregular metaphyses and poorly developed epiphyses.
• Others, e.g. acute cardiomyopathy and communicating hydrocephalus in some cases.
TABLE 11.17: Classification of mucopolysaccharidoses
DS: Dermatan sulphate; HS: Heparan sulphate; KS: Keratan sulphate; HSM: Hepatosplenomegaly; MR: Mental retardation; CC: Corneal clouding *Abnormal facial appearance.
**Dysostosis multiplex.
Fig. 11.10: Hurler syndrome: (A) Clinical appearance, note corneal clouding (inset); (B) beaking of the vertebrae (arrow).
Most cases develop recurrent respiratory infections, noisy breathing or obstructive airway disease due to facial deformities and die by 10 years.
Diagnosis rests on clinical and radiological findings with urinary excretion of dermatan and heparin sulfate. Definitive diagnosis requires molecular testing though specific enzyme assay in serum leukocytes or skin fibroblasts may be used. Prenatal diagnosis is possible using amniocytes or chorionic villous cells.
Treatment is supportive with control of cardiorespiratory complications, orthopedic corrections and hearing support. Although bone marrow transplant in severe phenotypes has shown significant clinical improvement in somatic features, skeletal, ocular and mental abnormalities are irreversible.
Enzyme replacement therapy (IV Laronidase every 2 weeks) reduces the GAG excretion and causes subjective improvement in respiratory symptoms. However, there is not much improvement in skeletal features and cardiac features.
Scheie syndrome (MPS IS) is mildest variant of MPS with onset beyond 5 years of age. These cases have normal stature and intelligence with minimum facial and skeletal features, usually characterized by: (a) joint stiffness, (b) severe corneal clouding, and (c) aortic valve disease.
Hunter syndrome (MPS II) is only X-linked recessive MPS, with two clinical variants—a severe form analogous to Hurler syndrome, and a mild form analogous to Scheie syndrome, except: (a) absence of corneal clouding, and (b) slower progression of somatic and neurological deterioration. Communicating hydrocephalus is common in severe form. Enzyme replacement therapy (IV Idursulfase every week) is available.
Sanfilippo syndrome (MPS III) includes four biochemical diverse but clinically similar types of MPS (A-D), each with different enzyme deficiency.
These cases usually present at 2-6 years of age with slowly progressive neurological impairment but only mild somatic disease (coarse hair, hirsutism, mild hepatosplenomegaly and skeletal features). Short stature and corneal clouding is rare. Severe neurological deterioration develops by 6-10 years, with death in adolescence.Morquio syndrome (MPS IV) is characterized by normal intelligence but severe skeletal deformities, i.e. (a) short trunkshort stature due to platyspondyly, i.e. flat vertebrae, (b) short neck with unstable odontoid process, susceptible for atlantoaxial subluxation, (c) kyphoscoliosis with dysplastic hips and flat feet, leading to waddling gait. Joint laxity, mild corneal clouding, hepatomegaly, cardiac valvular lesions and abnormal dentition may be present. Most cases survive till 3-4th decade of life.
Treatment involves enzyme replacement therapy (IV Elosulfase) and supportive care with clinical and neuroimaging monitoring for cervical cord compression and immediate neurosurgical intervention.
Maroteaux-Lamy syndrome (MPS VI), in severe form resembles Hurler syndrome, except absence of developmental delay. However, milder variants may be easily confused with Scheie syndrome. Spinal cord compression due to thickened cervical dura matter is common in mild forms. Enzyme replacement therapy (IV Galsulfase) is available.
Sly syndrome (MPS VII) is the rarest MPS with extremely variable presentation, ranging from hydrop fetalis or severe dysostosis multiplex in newborns, to features resembling Hurler syndrome in older children.
11.6.4