PORPHYRIA
Heme, composed of ferrous iron and protoporphyrins, is an essential constituent of hemoglobin, myoglobin and many other enzymes. The porphyrias are inherited disorders of heme biosynthesis due to various enzyme defects (Fig.
11.11), leading to abnormal tissue accumulation and urinary/fecal excretion of porphyrins or their precursors. Cutaneous photosensitivity and neurological involvement are predominant manifestations of porphyrin toxicity, apart from visceral features in some cases.Porphyrias are classified according to the order of defective enzyme in heme biosynthesis (Table 11.18) or the site of expression, i.e. acute hepatic porphyrias (ADP, AIP, HCP, VP) or erythrocytic porphyrias (CEP, PCT, HEP, EPP). Of these, most common are acute intermittent porphyria, porphyria cutaneous tarda and erythrocytic porphyria, discussed here in greater details.
Acute intermittent porphyria (AIP), is the commonest genetic porphyria, which usually presents beyond puberty.
Clinically, it present with acute episodes of: (a) severe abdominal pain with/without vomiting, mimicking surgical abdomen, (b) neuropathy with proximal muscle weakness and cranial nerve involvements, and (c) cardiovascular signs, e.g. tachycardia with hypertension,
Fig. 11.11: Important steps in heme synthesis and enzymatic origin of various porphyrias.
which may persist even after the episode. Attacks are precipitated by fasting, infections, surgery, drugs/ chemical ingestion or hormonal factors, e.g. menses in females. Skin manifestations are absent.
Diagnosis rests on decreased porphobilinogen deaminase activity in erythrocytes, though elevated levels of porphobilinogen in urine on Watson-Schwartz screening test or chromatographic studies are highly suggestive.
Treatment is largely supportive during the attack, which may be prevented by adequate caloric intake, avoidance of precipitant drugs and prompt management of infections. IV hematin (4 mg / kg / BD) may curtail severe attacks.
Porphyria cutanea tarda (PCT), the commonest porphyria, which may be inherited (autosomal dominant) or acquired. Acquired PCT is mainly seen in adults after exposure to factors, e.g. alcohol, estrogens or drugs.
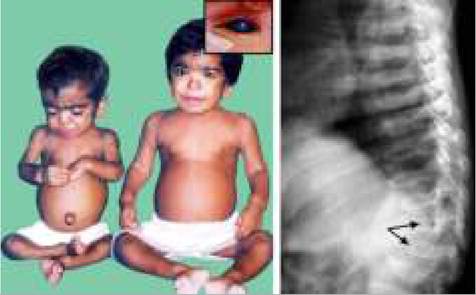
Clinically, cutaneous photosensitivity with formation of vesicles over exposed parts of body is pathognomonic of PCT, which may heal with crusting, scarring and residual pigmentation. Neurological or visceral manifestation are absent.
Diagnosis rests on elevated excretion of urinary and fecal porphyrins, specially isocoproporphyrin levels in stools. Daily porphyrin excretion in stools exceeds that in urine.
(EPP)
AP: Abdominal pain, HA: Hemolytic anemia, MOI: Modes of inheritance
Treatment is symptomatic, though avoidance of sunlight and precipitating factors in acquired disease is necessary.
Erythrocytic protoporphyria (EPP), an autosomal dominant ferrochelatase deficiency, typically manifests in childhood, with extreme cutaneous photosensitivity. Clinically, EPP present with stingy or burning sensations within 1-2 hours of sun exposure followed by erythema and edema. Vesicles are less common but repeated attacks lead to hyperkeratosis of exposed skin and onycholysis. Chronic hepatic disease and gallstones are common. Neuro-visceral involvement is absent.
Diagnosis rests on elevated free protoporphyrin levels in erythrocytes, plasma and stools, with normal urinary porphyrins.
Treatment includes avoidance of sun-exposure and use of topical sunscreen agents to prevent skin burning. Oral administration of #946;-carotene increases photo-tolerance to some extent.
BIBLIOGRAPHY
1. Narayanan DL et al. Genomic Testing for Diagnosis of Genetic Disorders in Children: Chromosomal Microarray and Next-Generation Sequencing. Indian Pediatr. 2020;57: 549.
2. John ST. Consensus Statement of the Neurodevelopmental Pediatrics Chapter of Indian Academy of Pediatrics (IAP) on the Management of Children with Down Syndrome Indian Pediatr. 2023;60:298.
3. Narayanan DL et al. Understanding Exome Sequencing: Tips for the Pediatrician. Indian Pediatr. 2021;58:771.
4. Bijarnia-Mahay S et al. Testing Modalities for Inborn Errors of Metabolism - What a Clinician Needs to Know? Indian Pediatr. 2019;56:757.
5. Puri RD et al. Diagnosis and Management of Gaucher Disease in India - Consensus Guidelines of the Gaucher Disease Task Force of the Society for Indian Academy of Medical Genetics and the Indian Academy of Pediatrics Indian Pediatr. 2018;55:143.
More on the topic PORPHYRIA:
- PORPHYRIA
- I OSTEOPOROSIS ^470 ^485 ^508 ^571
- 41 Hyperandrogenism
- Czechoslovakia
- Acknowledgements
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025