Myopathies
Polymyositis/Dermatomyositis
Polymyositis/dermatomyositis has been described in children ranging in age from infancy to adulthood. Children may result with progressive proximal muscle weakness, dysphagia due to involvement of pharyngeal musculature, dyspnea, and muscle tenderness.
A classic skin rash may or may not be present. Creatinine kinase values are often markedly elevated. Classic EMG findings include increased insertional activity with complex repetitive discharges; fibrillations and positive sharp waves; and low-amplitude, polyphasic, short-duration motor unit action potentials recruited rapidly in relation to the strength of contraction.Congenital Myopathies
Congenital myopathies are a heterogeneous group of disorders usually presenting with infantile hypotonia, normal cognitive status, and primary structural abnormalities of the muscle fibers, which are elucidated on histologic and electron microscopic evaluations of muscle biopsy specimens. Patients usually develop proximal greater than distal muscle weakness that is nonprogressive and static. These myopathies are described in the chapter on pediatric neuromuscular diseases. Nerve conduction studies are generally normal; however, there may be mild reductions in CMAP amplitudes. On needle EMG, findings are either normal or there may be mild, nonspecific changes, usually of a myopathic character (small-amplitude, short-duration polyphasic MUAPs). The only congenital myopathy consistently associated with abnormal spontaneous rest activity is myotubular (centronu- clear) myopathy. In this disorder, the EMG reveals myopathic motor unit action potentials with frequent complex repetitive discharges and diffuse fibrillation potentials.
Dystrophic Myopathies
The dystrophic myopathies are extensively described in the chapter on pediatric neuromuscular diseases. EMG is rarely used at the present for the diagnostic evaluation of a suspected dystrophic myopathy due to molecular genetic testing and the importance of muscle biopsy in differentiating among Duchenne muscular dystrophy, Becker muscular dystrophy, and limb girdle muscular dystrophies. EMG in dystrophic myopathies is characterized by low-amplitude, short-duration poly- phasic MUAPs (Fig.
7.14). Recruitment is myopathic in nature with increased recruitment or “early” recruitment demonstrated with slight effort. Interference pattern is usually full. Complex repetitive discharges (Fig. 7.15) and abnormal spontaneous rest activity may be present, reflecting membrane instability.Metabolic Myopathies
Nonspecific myopathic EMG findings may be demonstrated in metabolic myopathies. For example, absent maltase deficiency shows increased insertional activity; complex repetitive discharges; low-amplitude, short-duration MUAPs; profuse fibrillations; and positive sharp waves. Carnitine deficiency, a disorder of lipid metabolism, demonstrates increased recruitment for effort, decreased amplitudes of MUAPs and occasional fibrillations. EMG may be normal in many metabolic myopathies, such as carnitine palmityl transferase deficiency.
Myotonic Disorders
Myotonic disorders such as myotonic muscular dystrophy and Schwartz-Jampel syndrome may show myotonic discharges with either positive sharp wave or fibrillation configuration and a waxing and waning firing frequency. The myotonic discharges are often described as exhibiting the sound of a “dive bomber.” There may be profuse fibrillations and positive sharp waves. MUAPs are often of low amplitude and short duration. There may be more involvement of distal musculature than proximal musculature in myotonic muscular dystrophy. Again, with a known family history of myotonic muscular dystrophy, confirmation of the diagnosis in an individual with classic clinical features can be expeditiously and cost-effectively confirmed in the EMG laboratory. However, clinical trials frequently require molecular genetic confirmation of myotonic muscular dystrophy (DM1 versus DM2 and other myotonic disorders). so EMG is becoming less utilized diagnostically.
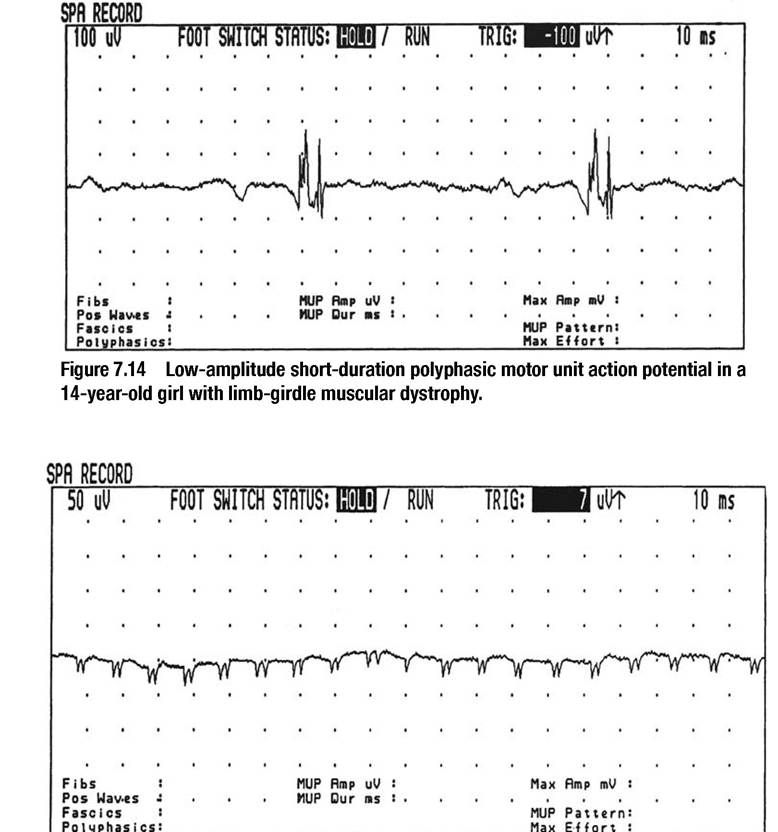
Figure 7.15 Complex repetitive discharges in a dystrophic myopathy.