SKELETAL MALIGNANCIES
Skeletal tumors are rare in first decade of life with peak incidence in adolescence, commonest being osteosarcoma followed by Ewing's sarcoma. Important similarities and differences between these tumors are given in Table 20.14.
Osteosarcoma arise from metaphyseal region of the rapidly growing long bones, e.g. distal femur or proximal tibia. Flat bones are rarely involved.
TABLE 20.14: Osteosarcoma vs Ewing's sarcoma
Osteosarcoma Ewing's sarcoma
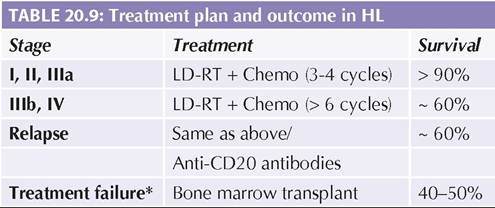
C: Chemotherapy; RT: Radiotherapy.
Histologically, these tumors comprise of highly malignant spindle cells, which produce extracellular osteoid. Epidemiology: These tumors are most common during physiological growth spurt in adolescents. Important high risk factors include: (a) hereditary retinoblastoma, (b) benign bony lesions, e.g., Paget's disease or Osteogenesis imperfecta, and (c) local radiotherapy for other malignancies. History of minor sport injury is often present. Germline mutations in tumor suppressor genes, e.g., RB1 or p53, are associated with higher risk of osteosarcoma.
Clinically most cases present with non-specific localized signs, e.g. pain, swelling, warmth and restricted movements, frequently misdiagnosed as inflammatory or traumatic lesions in early stages.
Diagnosis depends on biopsy. X-ray may reveal typical sun-ray or sun-burst appearance due to osteosclerosis with new bone formation (Fig. 20.3A). MRI is essential to ascertain the extent and operability of primary tumor along with chest CT and PET-scan to detect metastasis. Treatment includes: (a) preoperative chemotherapy (Doxorubicin, Cisplatin, Ifosfamide and Methotrexate) to shrink the tumor and improve the chances of limb salvage, followed by (b) surgical removal of tumor or amputation, and (c) post-operative chemotherapy.
Outcome: 5-year survival depends on extent of spread, ranging between ~60-70% in non-metastatic and lt;20% in metastatic disease.
Ewing sarcoma is a member of Ewing'sfamily—a group of small round-cell tumors of probably neural crest origin. Other members of this family include neuroblastoma, peripheral primitive neuro-ectodermal tumor (PPNET) and some soft-tissue sarcoma. Chromosomal translocations, e.g. t11:22 are common in Ewing's family of tumors.
Clinically, Ewing sarcoma commonly involves flat bones, e.g. pelvis, spine and chest wall (Askin tumor), presenting with local pain, tenderness and swelling. Some tumors may arise from diaphysis of long bones and soft tissues. Systemic manifestations, e.g., fever and weight loss are common.
Diagnosis depends on surgical biopsy. X-rays reveal typical 'onion-peel appearance' due to aggressive lytic lesions (Fig. 20.3B). Biopsy with immunohistochemistry (CD99, Vimentin) for neural cell markers is necessary to differentiate other round-cell tumors. Metastasis needs to be excluded by bone marrow, chest CT and PET-scan. Treatment includes preoperative chemotherapy with vincristine, cyclophosphamide, doxorubicin, etoposide and ifosfamide to shrink the tumor, followed by (a) surgical resection, and (b) postoperative chemotherapy.
While Ewing sarcoma is highly radio-sensitive and moderate-dose radiotherapy (5500-6000 cGy) is equally
Fig. 20.3A and B: (A) Sun-ray appearance in osteogenic sarcoma; (B) Onion-peel appearance in Ewing’s sarcoma.
effective, most centers prefer surgical resection for local tumor due to complications of radiotherapy on growing bones. However, radiotherapy is necessary for non- operable tumors.
Prognosis depends on the site and size of primary tumor as well as extent of metastasis, with 5-year survival ranging from 75% in small non-metastatic limb tumors to lt;20% in pelvic tumors or metastatic disease.
20.8 SOFT TISSUE SARCOMA
Sarcomas account for 6-7% of childhood tumors and include common rhabdomyosarcoma and uncommon nonrhabdomyosarcoma, e.g. fibrosarcoma, synovial sarcoma, etc.
Rhabdomyosarcoma accounts for ~4% of childhood cancers, more common in males and children with neurofibromatosis.
Pathologically, sarcoma are malignant tumors of primitive mesenchymal tissue and may develop at any site, usually head and neck (25%), genitourinary tract or extremities. Metastasis is common in lung and bones.
Histologically, these are similar to other round cell tumors, e.g. Ewing's sarcoma or neuroblastoma with differentiation based on immunohistochemistry and/ or molecular studies.
Three distinct histological types with differences in the treatment and outcome include-embryonal type (60%) with intermediate prognosis, alveolar type (25-40%) with worst prognosis and pleomorphic type, which is rare in childhood (lt; 1%).
Clinical presentation depends on the site of tumor and compression of neighboring structures. Common presentations include nasal congestion, epistaxis and dysphagia (nasopharyngeal tumors), cranial nerve palsies, headache/vomiting (intracranial tumors) proptosis and diminished vision (orbital tumors) and hematuria, constipation or incontinence (genitourinary
tumors). Vaginal RMS may present as a grapelike mass, coming through vagina (sarcoma botryoides).
Diagnosis rests on biopsy with immunochemistry, though CT-scan of the involved part is necessary to delineate tumor mass. Bone marrow studies, chest X-ray/ CT and PET scan are also required to detect metastasis. Cystourethrogram is indicated in genitourinary tumors. Treatment is multidisciplinary, based on location and stage of primary tumor. Completely resectable (Grade I) tumors are treated with surgery and post-operative chemotherapy, while those with regional lymph node involvement or partially resectability (Grade II/III) need surgery followed by chemotherapy as well as local radiotherapy. Grade IV tumors with distant metastasis are inoperable and treated with systemic chemotherapy and irradiation.
Prognosis: 5-year survival varies from ~90% in stage I/ II disease, to ~60% in advanced disease. Older children and metastatic disease have poor prognosis.
Non-rhabdomyosarcoma are rare in childhood except fibrosarcoma, which is the commonest soft-tissue sarcoma in infancy.
20.9
More on the topic SKELETAL MALIGNANCIES:
- Skeletal System
- Sarcoma
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025
- Chemotherapy
- 46 Cervical Cancer
- Ovary